Surfactant Protein A Impairs Genital HPV16 Pseudovirus Infection by Innate Immune Cell Activation in A Murine Model

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Binding of HPV16-PsVs to SP-A but Not SP-D Results in Increased Viral Uptake by RAW264.7 Macrophages
2.2. SP-A-Mediated HPV16-PsVs Uptake by RAW264.7 Macrophages Is Calcium-Dependent, but Not Dependent on the Lectin-Binding Domain
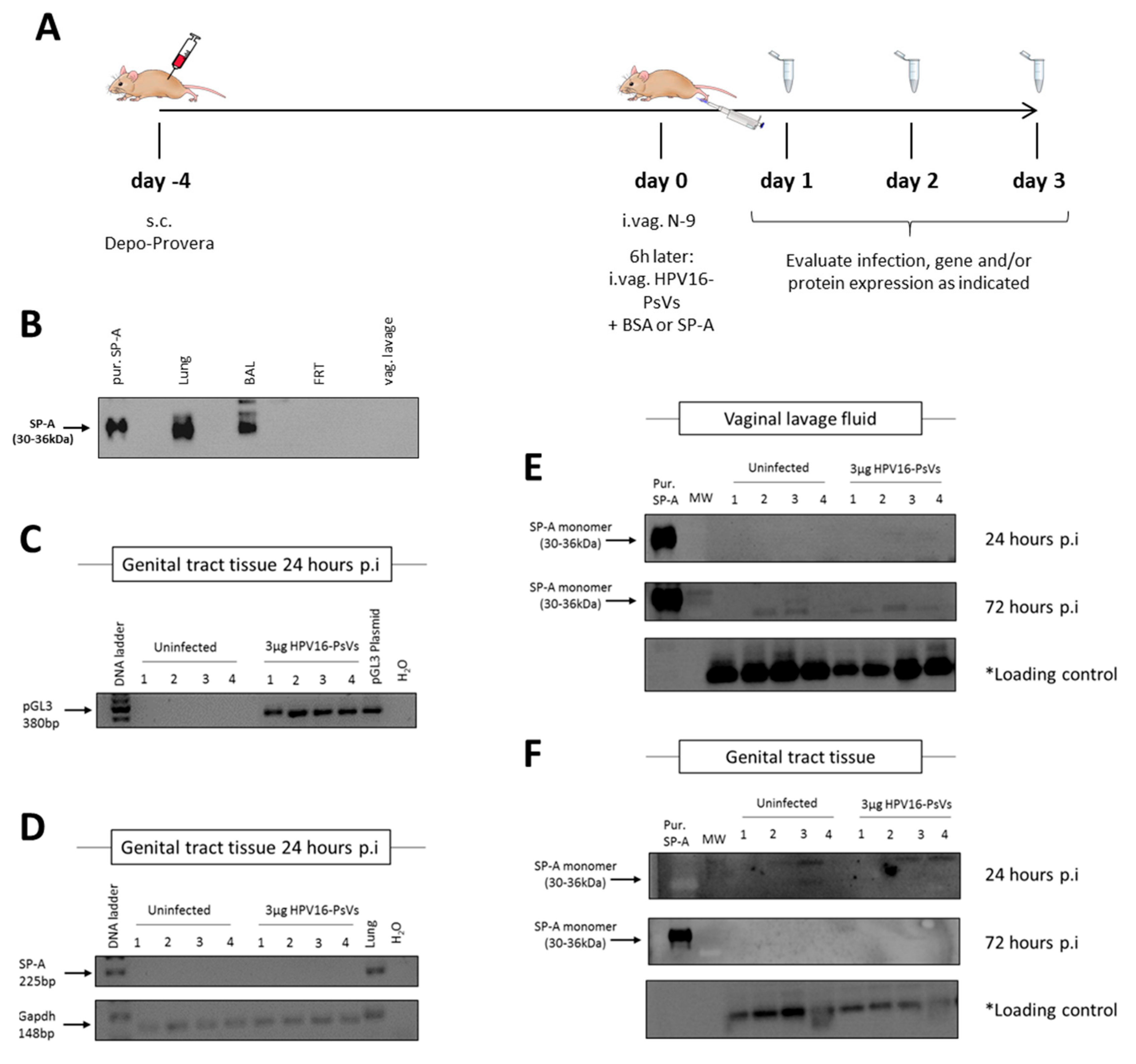
2.3. Cervicovaginal Challenge with HPV16-PsVs Does Not Alter SP-A Expression in the Murine FRT
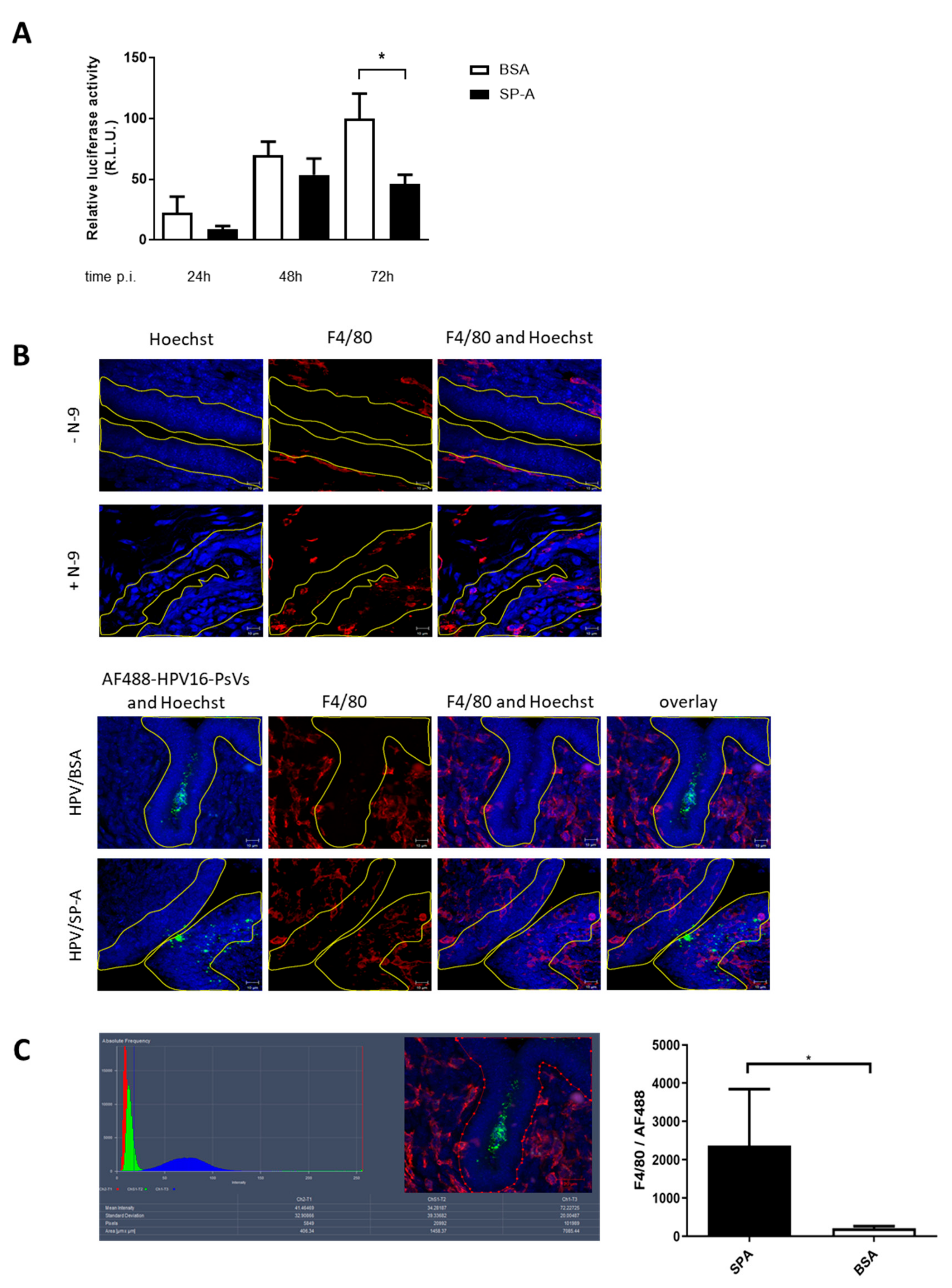
2.4. HPV16-PsVs Complexed with SP-A Reduces Infection by Increasing Macrophage Recruitment and Recognition by Innate Immune Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Purification of Native Human SP-A and Recombinant SP-D Proteins
4.3. HPV16 Pseudovirion Preparation, Viral Internalisation and Infection Assays
4.4. Co-Immunoprecipitation and Western Blotting
4.5. HPV16-PsVs Murine Cervicovaginal Challenge Model
4.6. Gene Expression Analysis
4.7. Immunohistochemistry
4.8. Flow Cytometry Analysis of Genital Tract Immune Cell Populations
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schäfer, G.; Blumenthal, M.J.; Katz, A.A. Interaction of human tumor viruses with host cell surface receptors and cell entry. Viruses 2015, 7, 2592–2617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- De Vuyst, H.; Alemany, L.; Lacey, C.; Chibwesha, C.J.; Sahasrabuddhe, V.; Banura, C.; Denny, L.; Parham, G.P. The burden of human papillomavirus infections and related diseases in sub-saharan africa. Vaccine 2013, 31 (Suppl. S5), F32–F46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, A.L. The interaction between human immunodeficiency virus and human papillomaviruses in heterosexuals in africa. J. Clin. Med. 2015, 4, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Denny, L. Control of cancer of the cervix in low- and middle-income countries. Ann. Surg. Oncol. 2015, 22, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Thompson, C.D.; Roberts, J.N.; Muller, M.; Lowy, D.R.; Schiller, J.T. Carrageenan is a potent inhibitor of papillomavirus infection. PLoS Pathog. 2006, 2, e69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, J.N.; Buck, C.B.; Thompson, C.D.; Kines, R.; Bernardo, M.; Choyke, P.L.; Lowy, D.R.; Schiller, J.T. Genital transmission of hpv in a mouse model is potentiated by nonoxynol-9 and inhibited by carrageenan. Nat. Med. 2007, 13, 857–861. [Google Scholar] [CrossRef] [Green Version]
- Marais, D.; Gawarecki, D.; Allan, B.; Ahmed, K.; Altini, L.; Cassim, N.; Gopolang, F.; Hoffman, M.; Ramjee, G.; Williamson, A.L. The effectiveness of carraguard, a vaginal microbicide, in protecting women against high-risk human papillomavirus infection. Antivir. Ther. 2011, 16, 1219–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Romero, J.A.; Abraham, C.J.; Rodriguez, A.; Kizima, L.; Jean-Pierre, N.; Menon, R.; Begay, O.; Seidor, S.; Ford, B.E.; Gil, P.I.; et al. Zinc acetate/carrageenan gels exhibit potent activity in vivo against high-dose herpes simplex virus 2 vaginal and rectal challenge. Antimicrob. Agents Chemother. 2012, 56, 358–368. [Google Scholar] [CrossRef] [Green Version]
- Skoler-Karpoff, S.; Ramjee, G.; Ahmed, K.; Altini, L.; Plagianos, M.G.; Friedland, B.; Govender, S.; De Kock, A.; Cassim, N.; Palanee, T.; et al. Efficacy of carraguard for prevention of hiv infection in women in South Africa: A randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 1977–1987. [Google Scholar] [CrossRef]
- Stanley, M. Hpv—Immune response to infection and vaccination. Infect. Agents Cancer 2010, 5, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, A.; Phipps, M.J.S.; Clark, H.W.; Skylaris, C.K.; Madsen, J. Surfactant proteins a and d: Trimerized innate immunity proteins with an affinity for viral fusion proteins. J. Innate Immun. 2019, 11, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, J.S.; Martin, J.L.; Azad, A.K.; McCarthy, T.R.; Kang, P.B.; Voelker, D.R.; Crouch, E.C.; Schlesinger, L.S. Surfactant protein d increases fusion of mycobacterium tuberculosis-containing phagosomes with lysosomes in human macrophages. Infect. Immun. 2006, 74, 7005–7009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaynor, C.D.; McCormack, F.X.; Voelker, D.R.; McGowan, S.E.; Schlesinger, L.S. Pulmonary surfactant protein a mediates enhanced phagocytosis of mycobacterium tuberculosis by a direct interaction with human macrophages. J. Immunol. 1995, 155, 5343–5351. [Google Scholar] [PubMed]
- Ghildyal, R.; Hartley, C.; Varrasso, A.; Meanger, J.; Voelker, D.R.; Anders, E.M.; Mills, J. Surfactant protein a binds to the fusion glycoprotein of respiratory syncytial virus and neutralizes virion infectivity. J. Infect. Dis. 1999, 180, 2009–2013. [Google Scholar] [CrossRef]
- Hartshorn, K.L.; White, M.R.; Shepherd, V.; Reid, K.; Jensenius, J.C.; Crouch, E.C. Mechanisms of anti-influenza activity of surfactant proteins a and d: Comparison with serum collectins. Am. J. Physiol. 1997, 273, L1156–L1166. [Google Scholar] [CrossRef]
- LeVine, A.M.; Elliott, J.; Whitsett, J.A.; Srikiatkhachorn, A.; Crouch, E.; DeSilva, N.; Korfhagen, T. Surfactant protein-d enhances phagocytosis and pulmonary clearance of respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2004, 31, 193–199. [Google Scholar] [CrossRef]
- LeVine, A.M.; Whitsett, J.A.; Gwozdz, J.A.; Richardson, T.R.; Fisher, J.H.; Burhans, M.S.; Korfhagen, T.R. Distinct effects of surfactant protein a or d deficiency during bacterial infection on the lung. J. Immunol. 2000, 165, 3934–3940. [Google Scholar] [CrossRef] [Green Version]
- LeVine, A.M.; Whitsett, J.A.; Hartshorn, K.L.; Crouch, E.C.; Korfhagen, T.R. Surfactant protein d enhances clearance of influenza a virus from the lung in vivo. J. Immunol. 2001, 167, 5868–5873. [Google Scholar] [CrossRef]
- Madan, T.; Eggleton, P.; Kishore, U.; Strong, P.; Aggrawal, S.S.; Sarma, P.U.; Reid, K.B. Binding of pulmonary surfactant proteins a and d to aspergillus fumigatus conidia enhances phagocytosis and killing by human neutrophils and alveolar macrophages. Infect. Immun. 1997, 65, 3171–3179. [Google Scholar] [PubMed]
- Thawer, S.; Auret, J.; Schnoeller, C.; Chetty, A.; Smith, K.; Darby, M.; Roberts, L.; Mackay, R.M.; Whitwell, H.J.; Timms, J.F.; et al. Surfactant protein-d is essential for immunity to helminth infection. PLoS Pathog. 2016, 12, e1005461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ujma, S.; Horsnell, W.G.; Katz, A.A.; Clark, H.W.; Schäfer, G. Non-pulmonary immune functions of surfactant proteins a and d. J. Innate Immun. 2017, 9, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Haczku, A. Protective role of the lung collectins surfactant protein a and surfactant protein d in airway inflammation. J. Allergy Clin. Immunol. 2008, 122, 861–879. [Google Scholar] [CrossRef] [Green Version]
- Kishore, U.; Greenhough, T.J.; Waters, P.; Shrive, A.K.; Ghai, R.; Kamran, M.F.; Bernal, A.L.; Reid, K.B.; Madan, T.; Chakraborty, T. Surfactant proteins sp-a and sp-d: Structure, function and receptors. Mol. Immunol. 2006, 43, 1293–1315. [Google Scholar] [CrossRef]
- Jäkel, A.; Qaseem, A.S.; Kishore, U.; Sim, R.B. Ligands and receptors of lung surfactant proteins sp-a and sp-d. Front. Biosci. 2013, 18, 1129–1140. [Google Scholar] [CrossRef]
- Wright, J.R. Immunoregulatory functions of surfactant proteins. Nat. Rev. Immunol. 2005, 5, 58–68. [Google Scholar] [CrossRef]
- Garcia-Verdugo, I.; Tanfin, Z.; Dallot, E.; Leroy, M.J.; Breuiller-Fouche, M. Surfactant protein a signaling pathways in human uterine smooth muscle cells. Biol. Reprod. 2008, 79, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Leth-Larsen, R.; Floridon, C.; Nielsen, O.; Holmskov, U. Surfactant protein d in the female genital tract. Mol. Hum. Reprod. 2004, 10, 149–154. [Google Scholar] [CrossRef]
- MacNeill, C.; Umstead, T.M.; Phelps, D.S.; Lin, Z.; Floros, J.; Shearer, D.A.; Weisz, J. Surfactant protein a, an innate immune factor, is expressed in the vaginal mucosa and is present in vaginal lavage fluid. Immunology 2004, 111, 91–99. [Google Scholar] [CrossRef]
- Madhukaran, S.P.; Kishore, U.; Jamil, K.; Choolani, M.; Lu, J. Decidual expression and localization of human surfactant protein sp-a and sp-d, and complement protein c1q. Mol. Immunol. 2015, 66, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Gaiha, G.D.; Dong, T.; Palaniyar, N.; Mitchell, D.A.; Reid, K.B.; Clark, H.W. Surfactant protein a binds to hiv and inhibits direct infection of cd4+ cells, but enhances dendritic cell-mediated viral transfer. J. Immunol. 2008, 181, 601–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, J.; Gaiha, G.D.; Palaniyar, N.; Dong, T.; Mitchell, D.A.; Clark, H.W. Surfactant protein d modulates hiv infection of both t-cells and dendritic cells. PLoS ONE 2013, 8, e59047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberley, R.E.; Goss, K.L.; Ault, K.A.; Crouch, E.C.; Snyder, J.M. Surfactant protein d is present in the human female reproductive tract and inhibits chlamydia trachomatis infection. Mol. Hum. Reprod. 2004, 10, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Day, P.M.; Kines, R.C. Current understanding of the mechanism of hpv infection. Gynecol. Oncol. 2010, 118, S12–S17. [Google Scholar] [CrossRef] [Green Version]
- Jakel, A.; Clark, H.; Reid, K.B.M.; Sim, R.B. The human lung surfactant proteins a (sp-a) and d (sp-d) interact with apoptotic target cells by different binding mechanisms. Immunobiology 2010, 215, 551–558. [Google Scholar] [CrossRef]
- Roberts, J.N. Infection of murine vaginal or endocervical mucosa with human papillomavirus pseudovirions. Nat. Protoc. 2007. [Google Scholar] [CrossRef]
- Shafti-Keramat, S.; Handisurya, A.; Kriehuber, E.; Meneguzzi, G.; Slupetzky, K.; Kirnbauer, R. Different heparan sulfate proteoglycans serve as cellular receptors for human papillomaviruses. J. Virol. 2003, 77, 13125–13135. [Google Scholar] [CrossRef] [Green Version]
- Alt, C.; Harrison, T.; Dousman, L.; Fujita, N.; Shew, K.; Tran, T.T.; Shayesteh, S.; Matsukawa, A.; Mirsalis, J.; D’Andrea, A. Increased ccl2 expression and macrophage/monocyte migration during microbicide-induced vaginal irritation. Curr. HIV Res. 2009, 7, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Okogbule-Wonodi, A.C.; Chesko, K.L.; Famuyide, M.E.; Viscardi, R.M. Surfactant protein-a enhances ureaplasmacidal activity in vitro. Innate Immun. 2011, 17, 145–151. [Google Scholar] [CrossRef]
- Oberley, R.E.; Ault, K.A.; Neff, T.L.; Khubchandani, K.R.; Crouch, E.C.; Snyder, J.M. Surfactant proteins a and d enhance the phagocytosis of chlamydia into thp-1 cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L296–L306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, J.F.; Mealy, T.R.; McCormack, F.X.; Seaton, B.A. Crystal structure of trimeric carbohydrate recognition and neck domains of surfactant protein a. J. Biol. Chem. 2003, 278, 43254–43260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, J.; Takahashi, M.; Saito, A.; Murata, M.; Kurimura, Y.; Nishitani, C.; Takamiya, R.; Uehara, Y.; Hasegawa, Y.; Hiyama, Y.; et al. Surfactant protein a inhibits growth and adherence of uropathogenic escherichia coli to protect the bladder from infection. J. Immunol. 2017, 198, 2898–2905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberley, R.E.; Goss, K.L.; Hoffmann, D.S.; Ault, K.A.; Neff, T.L.; Ramsey, K.H.; Snyder, J.M. Regulation of surfactant protein d in the mouse female reproductive tract in vivo. Mol. Hum. Reprod. 2007, 13, 863–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhukaran, S.P.; Koippallil Gopalakrishnan, A.R.; Pandit, H.; Marri, E.D.; Kouser, L.; Jamil, K.; Alhamlan, F.S.; Kishore, U.; Madan, T. Expression of surfactant proteins sp-a and sp-d in murine decidua and immunomodulatory effects on decidual macrophages. Immunobiology 2016, 221, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Fortes, H.R.; von Ranke, F.M.; Escuissato, D.L.; Araujo Neto, C.A.; Zanetti, G.; Hochhegger, B.; Souza, C.A.; Marchiori, E. Recurrent respiratory papillomatosis: A state-of-the-art review. Respir. Med. 2017, 126, 116–121. [Google Scholar] [CrossRef] [Green Version]
- Buck, C.B.; Pastrana, D.V.; Lowy, D.R.; Schiller, J.T. Generation of hpv pseudovirions using transfection and their use in neutralization assays. Methods Mol. Med. 2005, 119, 445–462. [Google Scholar]
- Watson, A.; Kronqvist, N.; Spalluto, C.M.; Griffiths, M.; Staples, K.J.; Wilkinson, T.; Holmskov, U.; Sorensen, G.L.; Rising, A.; Johansson, J.; et al. Novel expression of a functional trimeric fragment of human sp-a with efficacy in neutralisation of rsv. Immunobiology 2017, 222, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Clark, H.W.; Mackay, R.M.; Deadman, M.E.; Hood, D.W.; Madsen, J.; Moxon, E.R.; Townsend, J.P.; Reid, K.B.M.; Ahmed, A.; Shaw, A.J.; et al. Crystal structure of a complex of surfactant protein d (sp-d) and haemophilus influenzae lipopolysaccharide reveals shielding of core structures in sp-d-resistant strains. Infect. Immun. 2016, 84, 1585–1592. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, G.; Graham, L.M.; Lang, D.; Blumenthal, M.J.; Bergant Marusic, M.; Katz, A.A. Vimentin modulates infectious internalisation of hpv16 pseudovirions. J. Virol. 2017, 91, e00307-17. [Google Scholar] [CrossRef] [Green Version]
- Bergant Marusic, M.; Ozbun, M.A.; Campos, S.K.; Myers, M.P.; Banks, L. Human papillomavirus l2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 2012, 13, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Kaushic, C.; Ashkar, A.A.; Reid, L.A.; Rosenthal, K.L. Progesterone increases susceptibility and decreases immune responses to genital herpes infection. J. Virol. 2003, 77, 4558–4565. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ujma, S.; Carse, S.; Chetty, A.; Horsnell, W.; Clark, H.; Madsen, J.; Mackay, R.-M.; Watson, A.; Griffiths, M.; Katz, A.A.; et al. Surfactant Protein A Impairs Genital HPV16 Pseudovirus Infection by Innate Immune Cell Activation in A Murine Model. Pathogens 2019, 8, 288. https://doi.org/10.3390/pathogens8040288
Ujma S, Carse S, Chetty A, Horsnell W, Clark H, Madsen J, Mackay R-M, Watson A, Griffiths M, Katz AA, et al. Surfactant Protein A Impairs Genital HPV16 Pseudovirus Infection by Innate Immune Cell Activation in A Murine Model. Pathogens. 2019; 8(4):288. https://doi.org/10.3390/pathogens8040288
Chicago/Turabian StyleUjma, Sylvia, Sinead Carse, Alisha Chetty, William Horsnell, Howard Clark, Jens Madsen, Rose-Marie Mackay, Alastair Watson, Mark Griffiths, Arieh A. Katz, and et al. 2019. "Surfactant Protein A Impairs Genital HPV16 Pseudovirus Infection by Innate Immune Cell Activation in A Murine Model" Pathogens 8, no. 4: 288. https://doi.org/10.3390/pathogens8040288