
The Global Spread Pattern of Rat Lungworm Based on Mitochondrial Genetics

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mitochondrial Genomes
2.2. Mitochondrial Gene Sequences
2.3. Phylogenetic Analysis
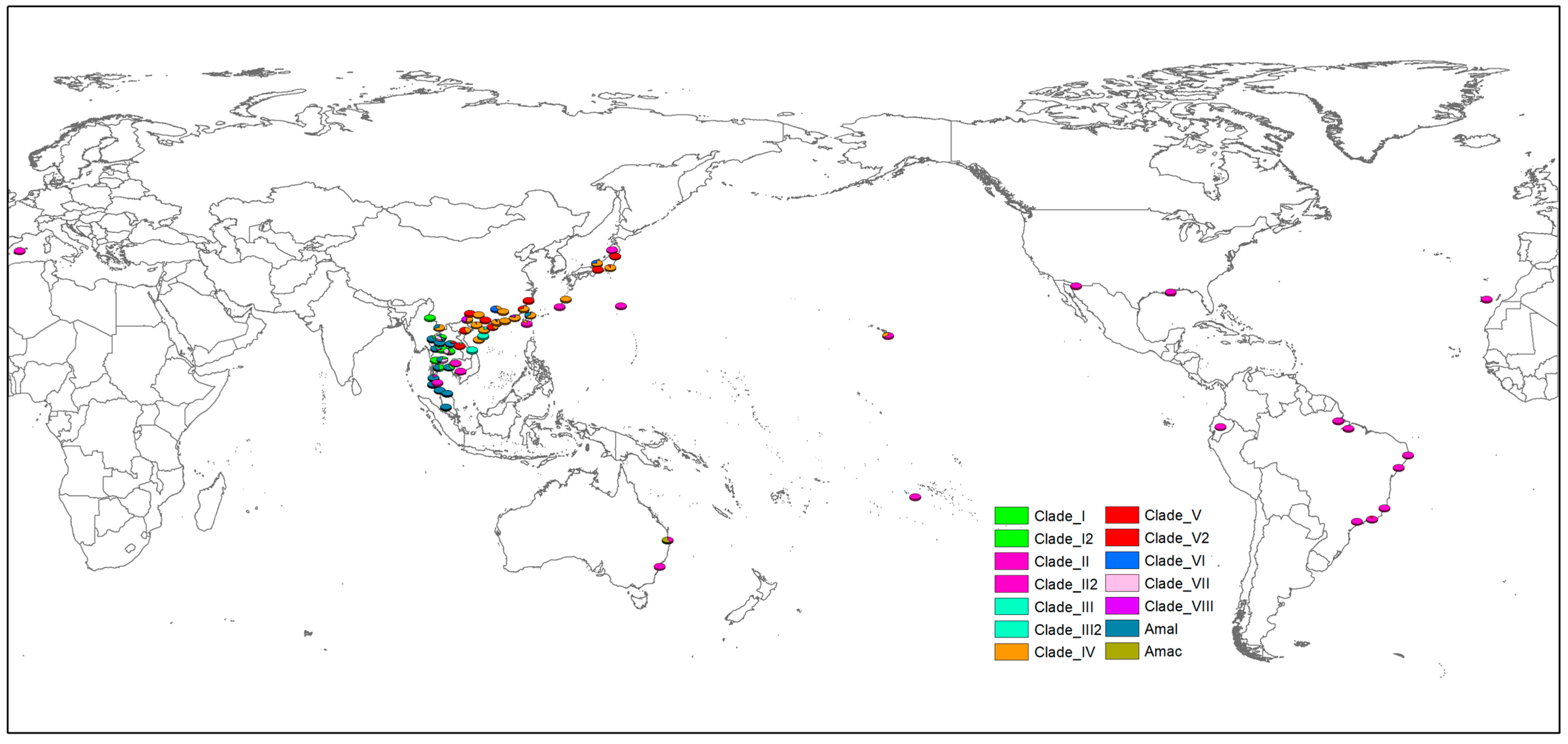
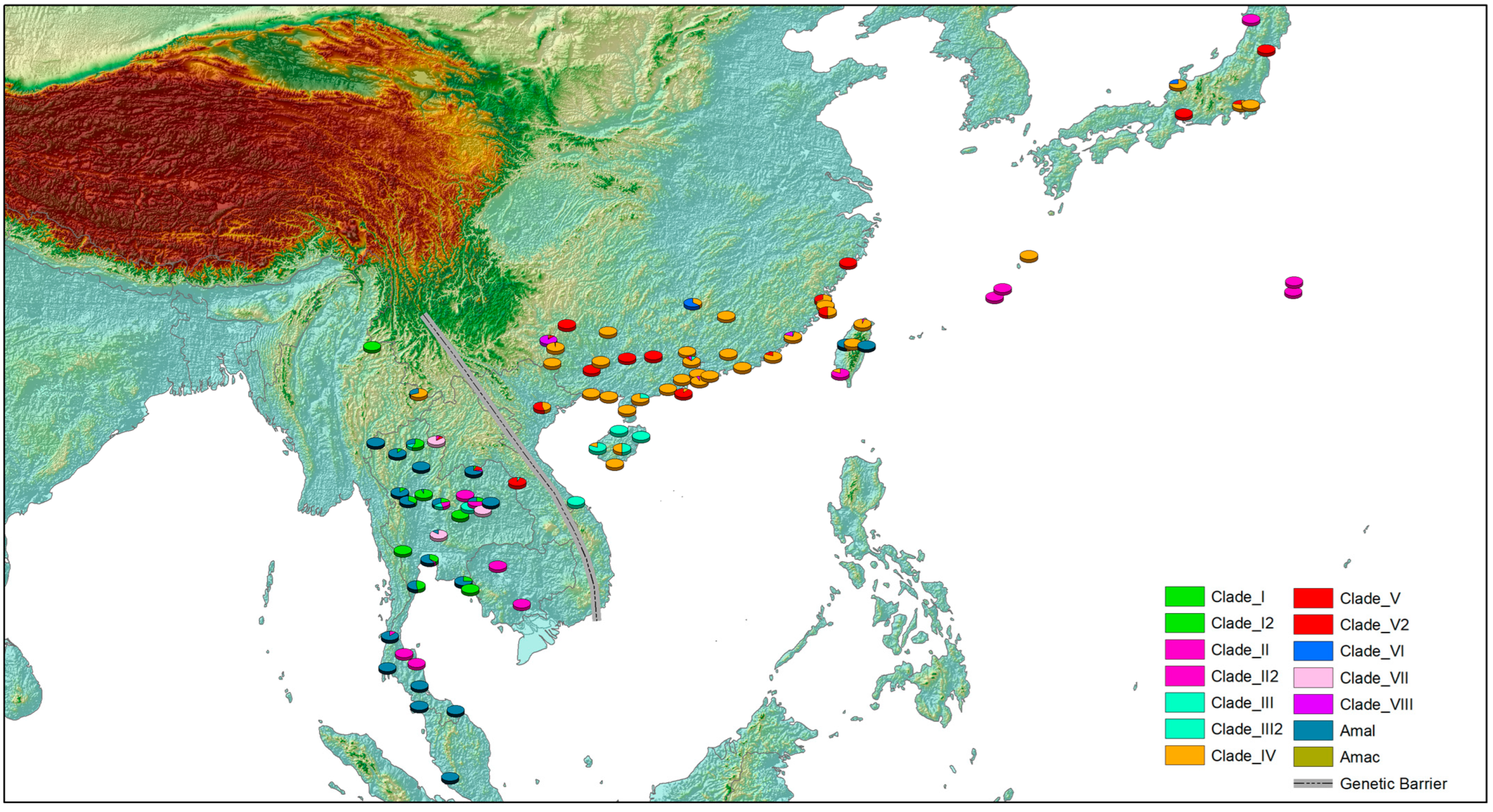
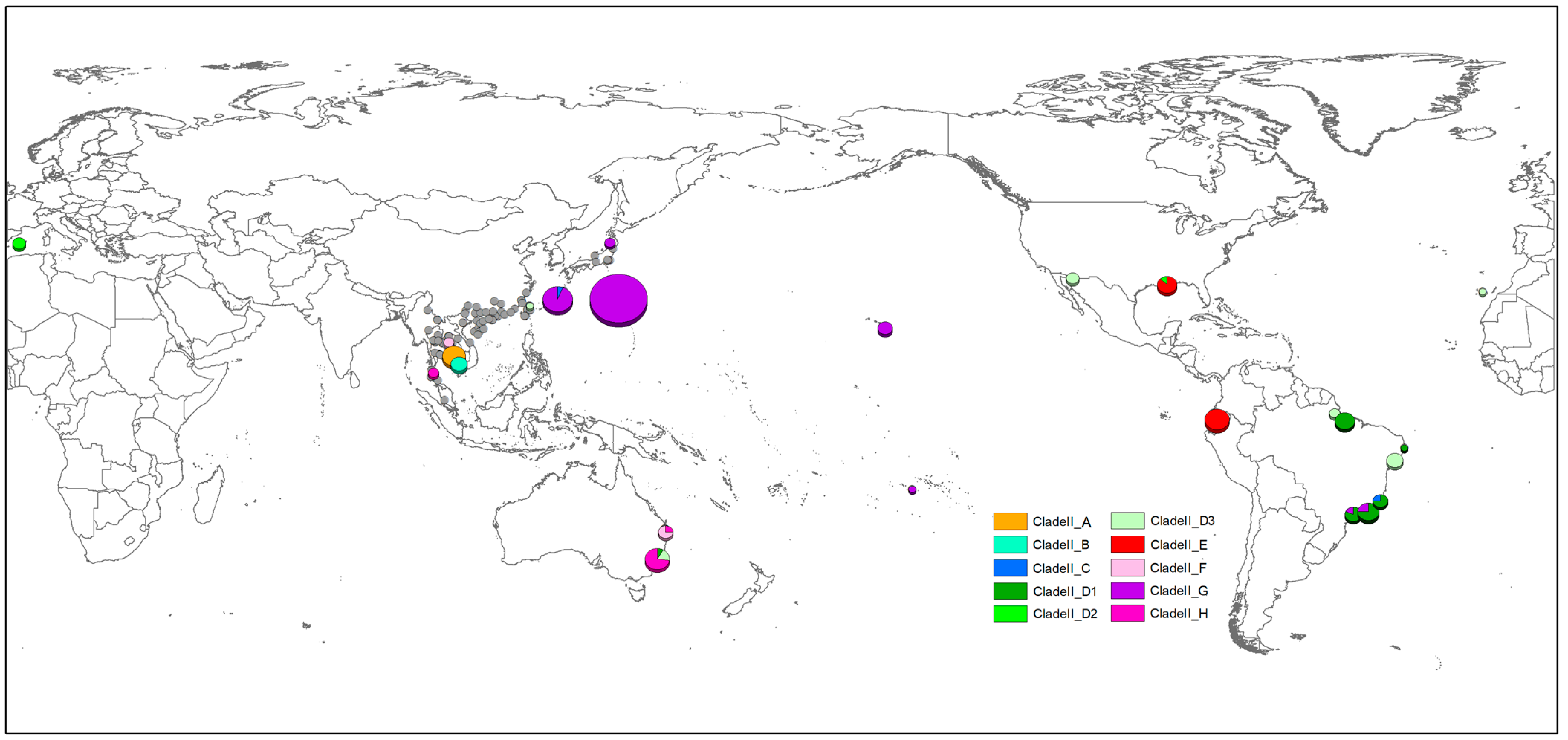
2.4. Mapping the Distribution Pattern
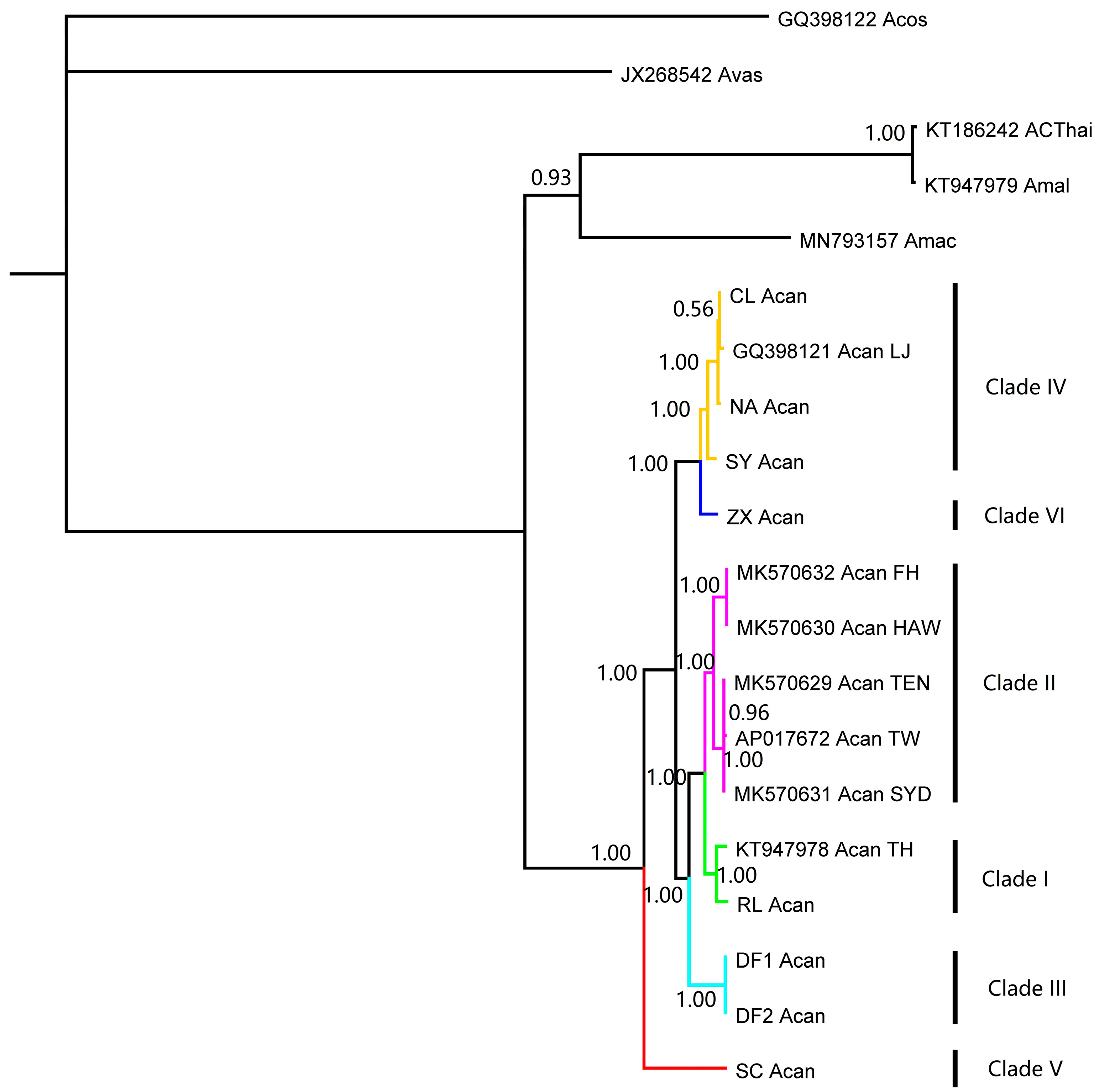
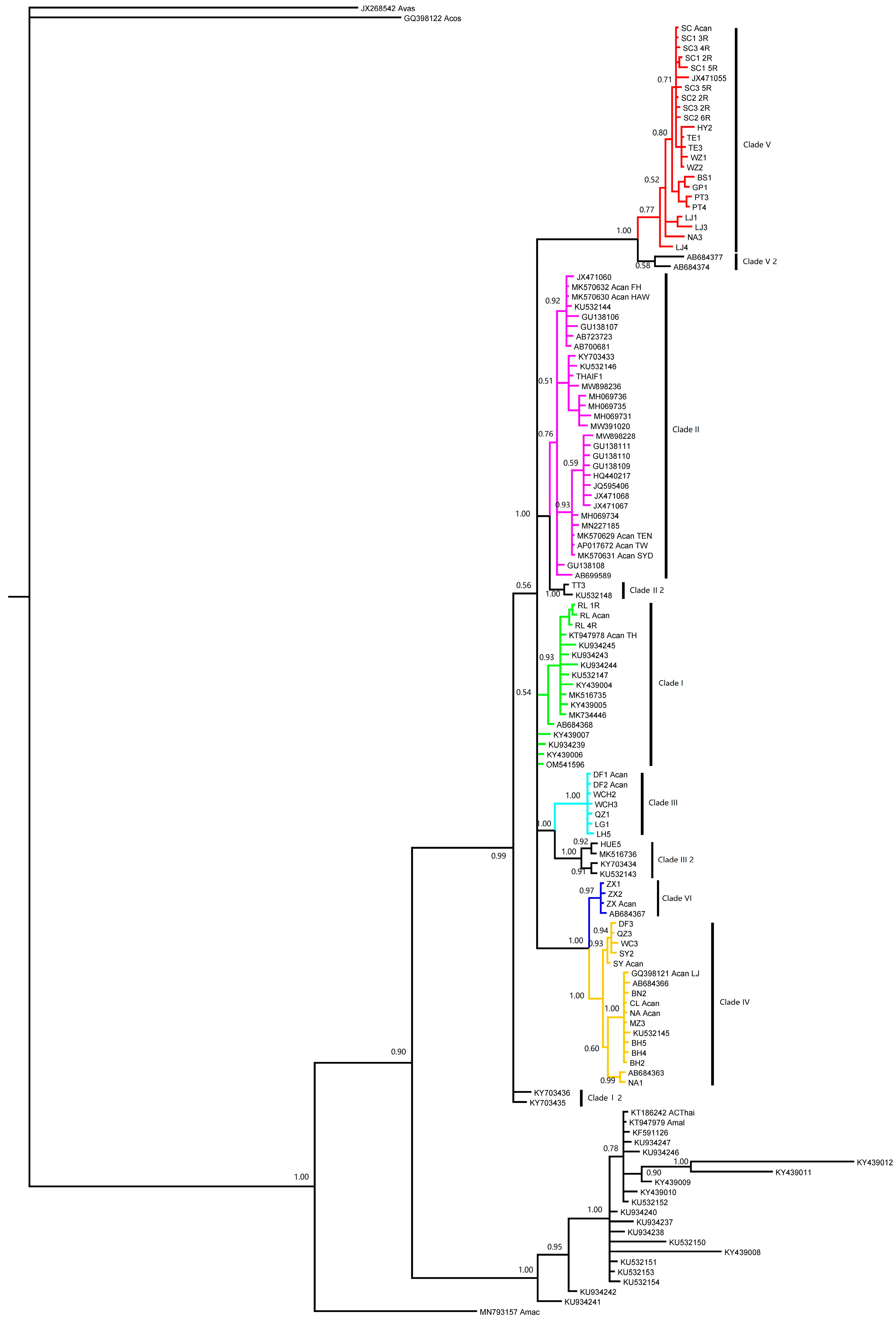
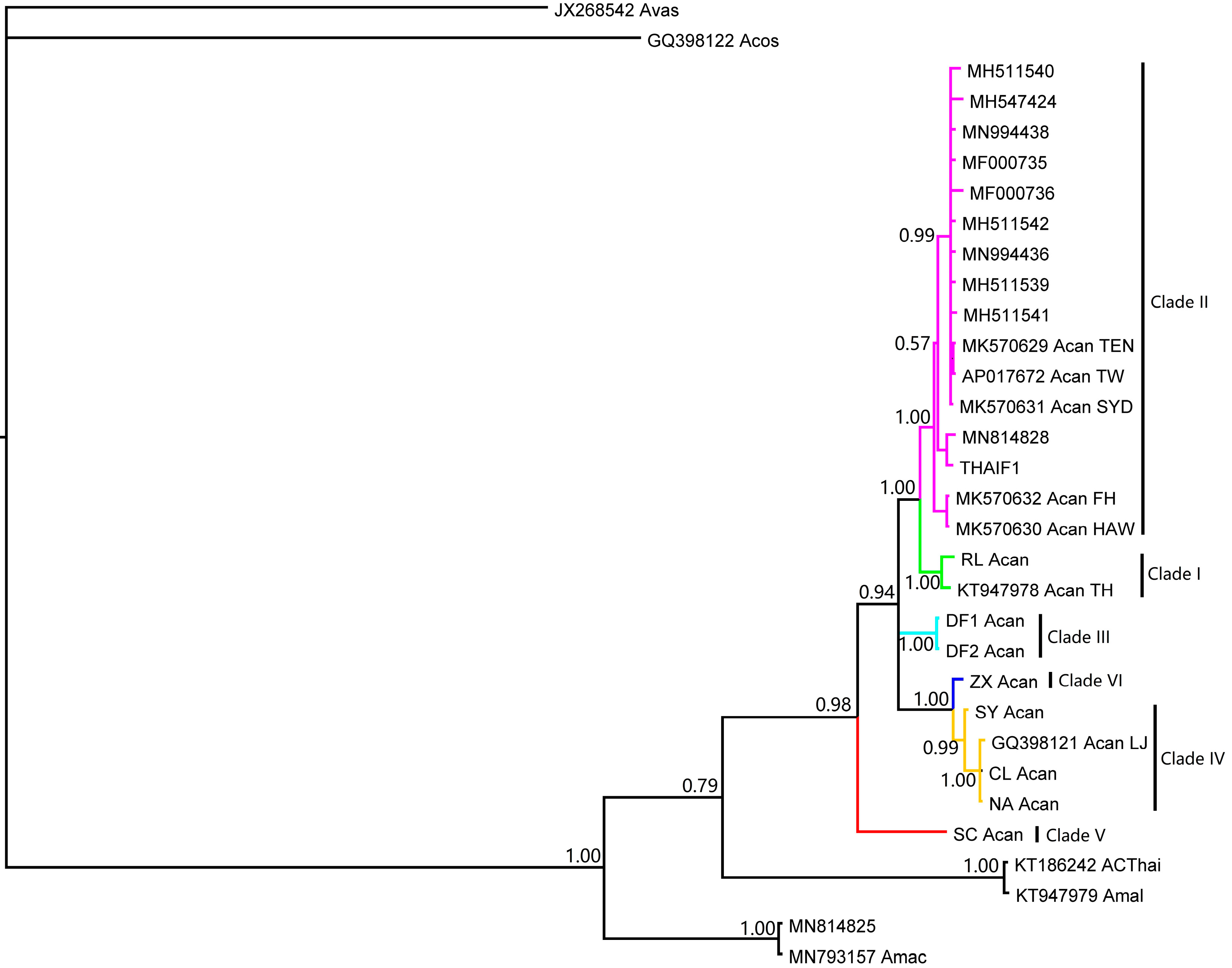
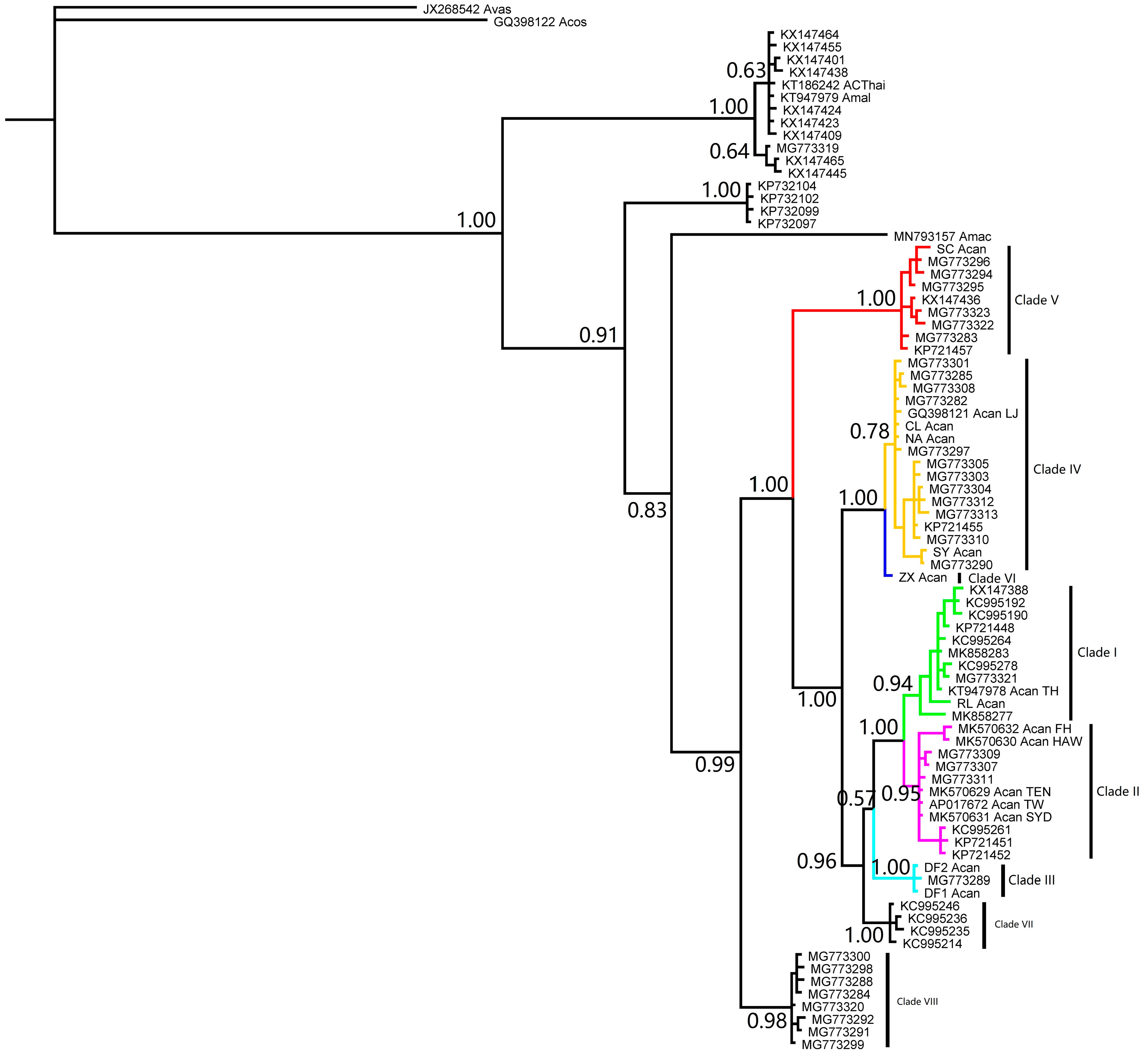
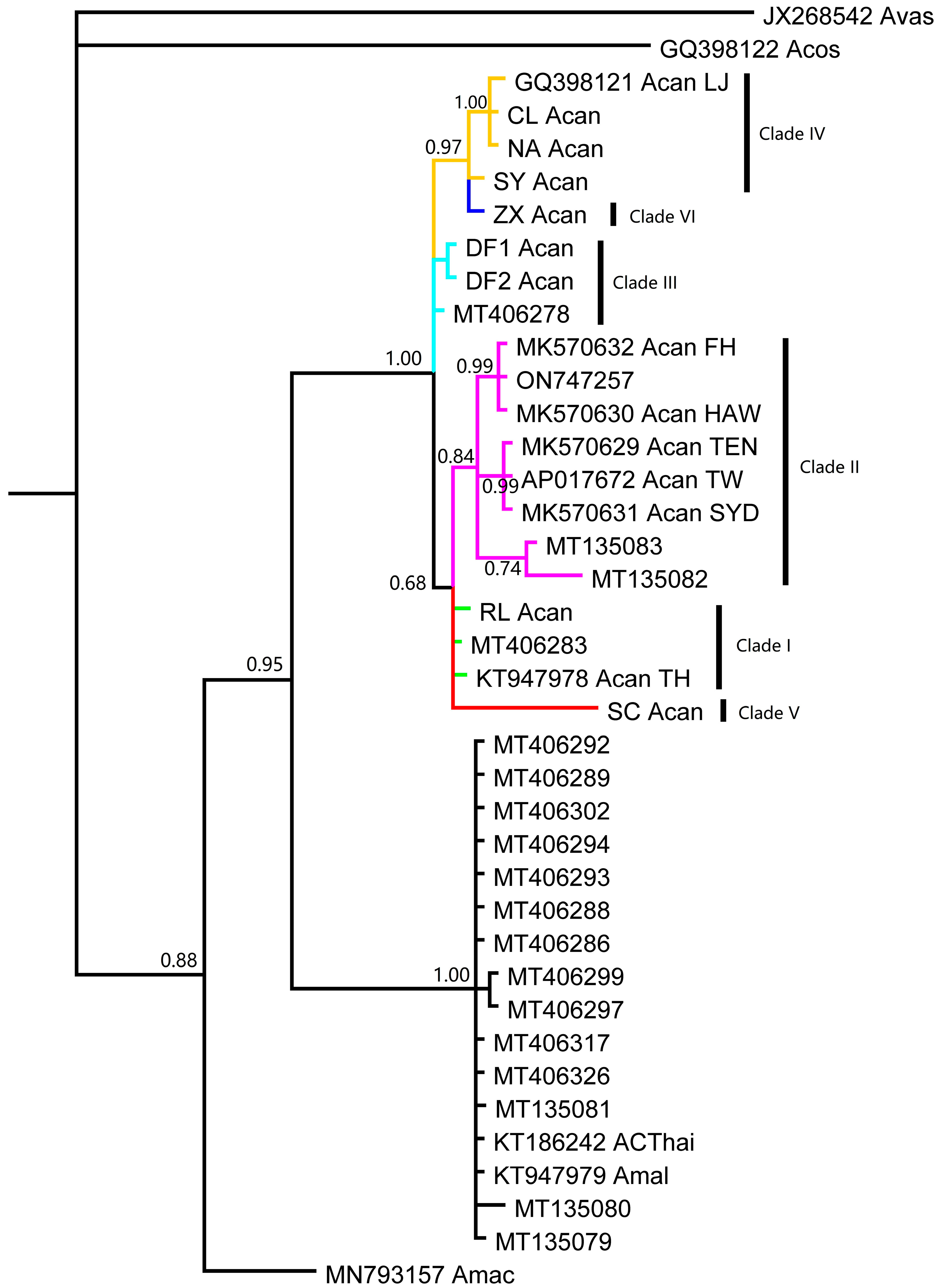
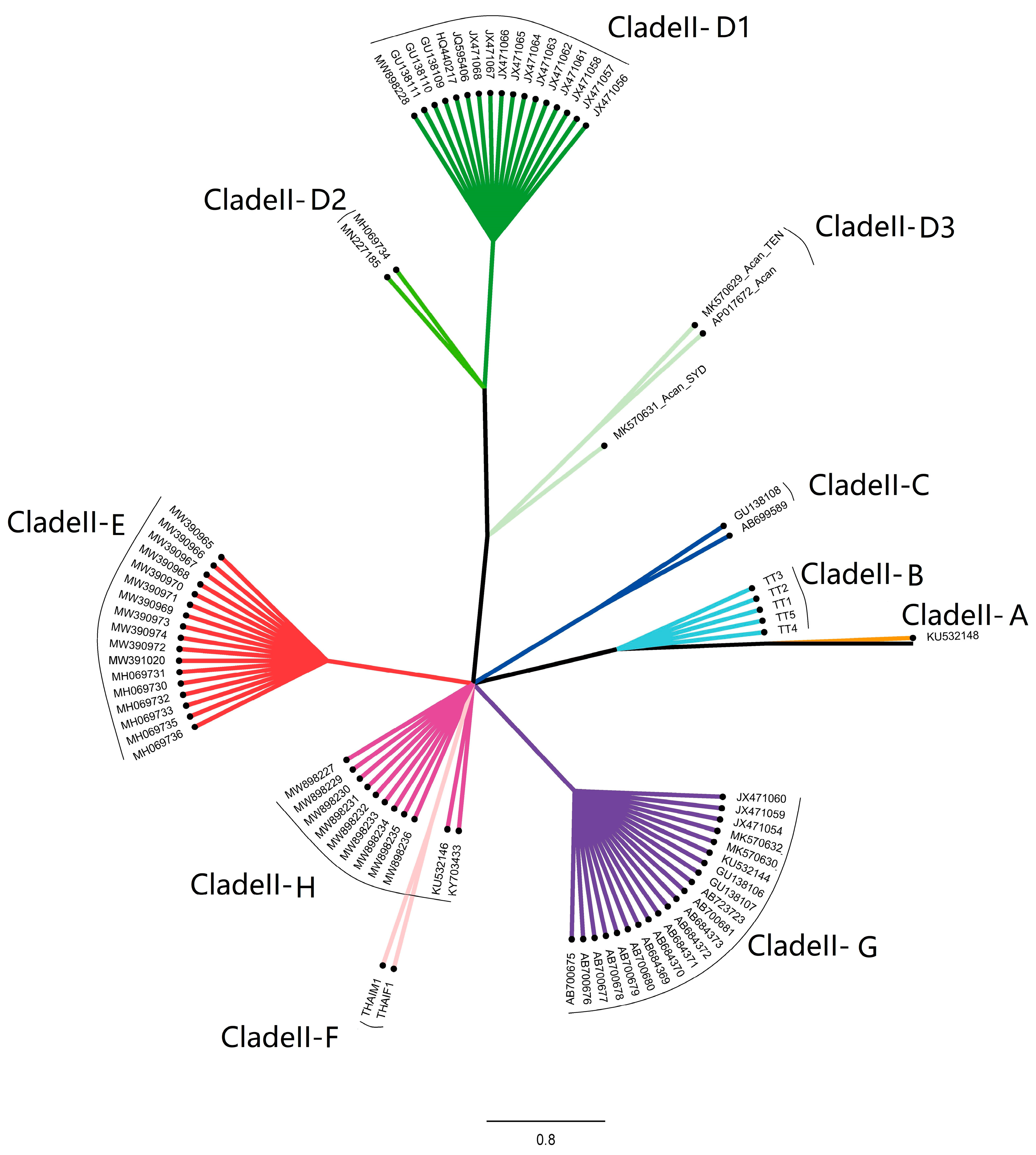
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lv, S.; Zhang, Y.; Steinmann, P.; Zhou, X.N.; Utzinger, J. Helminth infections of the central nervous system occurring in Southeast Asia and the Far East. Adv. Parasitol. 2010, 72, 351–408. [Google Scholar] [PubMed]
- Wang, Q.P.; Lai, D.H.; Zhu, X.Q.; Chen, X.G.; Lun, Z.R. Human angiostrongyliasis. Lancet Infect. Dis. 2008, 8, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Barratt, J.; Chan, D.; Sandaradura, I.; Malik, R.; Spielman, D.; Lee, R.; Marriott, D.; Harkness, J.; Ellis, J.; Stark, D. Angiostrongylus cantonensis: A review of its distribution, molecular biology and clinical significance as a human pathogen. Parasitology 2016, 143, 1087–1118. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.T. A preliminary report on a survey of animal parasites of Canton, China, rats. Lingnan Sci. J. 1933, 12, 65–74. [Google Scholar]
- Nomura, S.; Lin, P.H. First case report of human infection with Haemostrongylus ratti Yokogawa. Taiwan No Ikai 1945, 3, 589–592. [Google Scholar]
- Bailey, C.A. An Epidemic of Eosinophilic Meningitis, a Previously Undescribed Disease, Occuring on Ponape, Eastern Carolines; Report No. 7; Naval Medical Research Institute, National Naval Medical Center: Bethesda, MD, USA, 1948; pp. 1–23. [Google Scholar]
- Rosen, L.; Laigret, J.; Bories, S. Observations on an outbreak of eosinophilic meningitis on Tahiti, French Polynesia. Am. J. Hyg. 1961, 74, 26–42. [Google Scholar]
- Alicata, J.E.; Jindrak, K. Angiostrongylosis in the Pacific and Southeast Asia; Thomas Publisher: Springfield, IL, USA, 1970; p. 105. [Google Scholar]
- Solorzano Alava, L.F.; Martini Robles, L.; Hernandez Alvarez, H.; Sarracent Perez, J.; Muzzio Aroca, J.; Rojas Rivero, L. Angiostrongylus cantonensis: An emerging parasite in Ecuador. Rev. Cub. Med. Trop. 2014, 66, 20–33. [Google Scholar]
- Foronda, P.; Lopez-Gonzalez, M.; Miquel, J.; Torres, J.; Segovia, M.; Abreu-Acosta, N.; Casanova, J.C.; Valladares, B.; Mas-Coma, S.; Bargues, M.D.; et al. Finding of Parastrongylus cantonensis (Chen, 1935) in Rattus rattus in Tenerife, Canary Islands (Spain). Acta Trop. 2010, 114, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.H.; Moraes, C.; Almada, G.L.; Carvalho, O.S.; Caldeira, R.L.; Mendonça, C.L.; Zanon, D.M.; Antunes, F.A.; Santelli, A.C.; Castro, S.; et al. First reported outbreak of eosinophilic meningitis caused by Angiostrongylus cantonensis in Brazil. In Proceedings of the International Conference on Emerging Infectious Diseases, Atlanta, GA, USA, 16–19 March 2008; p. 59. [Google Scholar]
- Lowe, S.; Browne, M.; Boudjelas, S.; De Poorter, M. 100 of the World’s Worst Invasive Alien Species A selection from the Global Invasive Species Database; The Invasive Species Specialist Group (ISSG) a specialist group of the Species Survival Commission (SSC) of the World Conservation Union (IUCN): Auckland, New Zealand, 2000; p. 12. [Google Scholar]
- Kliks, M.M.; Palumbo, N.E. Eosinophilic meningitis beyond the Pacific Basin: The global dispersal of a peridomestic zoonosis caused by Angiostrongylus cantonensis, the nematode lungworm of rats. Soc. Sci. Med. 1992, 34, 199–212. [Google Scholar] [CrossRef]
- Mead, A.R. The Giant African Snail: A Problem in Economic Malacology; University of Chicago Press: Chicago, IL, USA, 1961; p. 257. [Google Scholar]
- Kliks, M.M.; Kroenke, K.; Hardman, J.M. Eosinophilic radiculomyeloencephalitis: An angiostrongyliasis outbreak in American Samoa related to ingestion of Achatina fulica snails. Am. J. Trop. Med. Hyg. 1982, 31, 1114–1122. [Google Scholar] [CrossRef]
- Graeff-Teixeira, C. Expansion of Achatina fulica in Brazil and potential increased risk for angiostrongyliasis. Trans. R. Soc. Trop. Med. Hyg. 2007, 101, 743–744. [Google Scholar] [CrossRef]
- Joshi, R.C.; Sebastian, L.S. Global Advances in Ecology and Management of Golden Apple Snails; PhilRice: Nueva Ecija, Philippines, 2006; p. 588. [Google Scholar]
- Lv, S.; Zhang, Y.; Steinmann, P.; Zhou, X.N. Emerging angiostrongyliasis in Mainland China. Emerg. Infect. Dis. 2008, 14, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Bhaibulaya, M.; Cross, J.H. Angiostrongylus malaysiensis (Nematoda: Metastrongylidae), a new species of rat lung-worm from Malaysia. Southeast Asian J. Trop. Med. Public Health 1971, 2, 527–533. [Google Scholar] [PubMed]
- Bhaibulaya, M. A new species of Angiostrongylus in an Australian rat, Rattus fuscipes. Parasitology 1968, 58, 789–799. [Google Scholar] [CrossRef]
- Prociv, P.; Spratt, D.M.; Carlisle, M.S. Neuro-angiostrongyliasis: Unresolved issues. Int. J. Parasitol. 2000, 30, 1295–1303. [Google Scholar] [CrossRef]
- Stafford, E.E.; Tanudjaja, S.; Purnomo; Carney, W.P. Angiostrongylus malaysiensis in Indonesia. Southeast Asian J. Trop. Med. Public Health 1976, 7, 490–491. [Google Scholar]
- Lim, B.L.; Ramachandran, C.P. Ecological studies on Angiostrongylus malaysiensis (Nematoda: Metastrongylidae) in Malaysia. In Studies on Angiostrongyliasis in Southeast Asia and Australia; John, H.C., Ed.; U. S. Naval Medical Research Unit No. 2: Taipei, Taiwan, 1979; p. 164. [Google Scholar]
- Watthanakulpanich, D.; Jakkul, W.; Chanapromma, C.; Ketboonlue, T.; Dekumyoy, P.; Lv, Z.; Chan, A.H.E.; Thaenkham, U.; Chaisiri, K. Co-occurrence of Angiostrongylus malaysiensis and Angiostrongylus cantonensis DNA in cerebrospinal fluid: Evidence from human eosinophilic meningitis after ingestion of raw snail dish in Thailand. Food Waterborne Parasitol. 2021, 24, e00128. [Google Scholar] [CrossRef]
- Mallaiyaraj Mahalingam, J.T.; Calvani, N.E.D.; Lee, R.; Malik, R.; Šlapeta, J. Using cerebrospinal fluid to confirm Angiostrongylus cantonensis as the cause of canine neuroangiostrongyliasis in Australia where A. cantonensis and Angiostrongylus mackerrasae co-exist. Curr. Res. Parasitol. Vector Borne Dis. 2021, 1, 100033. [Google Scholar] [CrossRef]
- Valentyne, H.; Spratt, D.M.; Aghazadeh, M.; Jones, M.K.; Šlapeta, J. The mitochondrial genome of Angiostrongylus mackerrasae is distinct from A. cantonensis and A. malaysiensis. Parasitology 2020, 147, 681–688. [Google Scholar] [CrossRef]
- Dumidae, A.; Janthu, P.; Subkrasae, C.; Dekumyoy, P.; Thanwisai, A.; Vitta, A. Genetic characterization of Angiostrongylus larvae and their intermediate host, Achatina fulica, in Thailand. PLoS ONE 2019, 14, e0223257. [Google Scholar] [CrossRef]
- Lv, S.; Zhang, Y.; Steinmann, P.; Utzinger, J.; Zhou, X.N. The genetic variation of Angiostrongylus cantonensis in the People’s Republic of China. Infect. Dis. Poverty 2017, 6, 125. [Google Scholar] [CrossRef] [Green Version]
- Jhan, K.Y.; Cheng, C.J.; Jung, S.M.; Lai, Y.J.; Chen, K.Y.; Wang, L.C. Co-Therapy of Albendazole and Dexamethasone Reduces Pathological Changes in the Cerebral Parenchyma of Th-1 and Th-2 Dominant Mice Heavily Infected with Angiostrongylus cantonensis: Histopathological and RNA-seq Analyses. Biomolecules 2021, 11, 536. [Google Scholar] [CrossRef]
- Červená, B.; Modrý, D.; Fecková, B.; Hrazdilová, K.; Foronda, P.; Alonso, A.M.; Lee, R.; Walker, J.; Niebuhr, C.N.; Malik, R.; et al. Low diversity of Angiostrongylus cantonensis complete mitochondrial DNA sequences from Australia, Hawaii, French Polynesia and the Canary Islands revealed using whole genome next-generation sequencing. Parasit Vectors 2019, 12, 241. [Google Scholar] [CrossRef] [Green Version]
- Lv, S.; Zhang, Y.; Liu, H.X.; Hu, L.; Yang, K.; Steinmann, P.; Chen, Z.; Wang, L.Y.; Utzinger, J.; Zhou, X.N. Invasive snails and an emerging infectious disease: Results from the first national survey on Angiostrongylus cantonensis in China. PLoS Negl. Trop. Dis. 2009, 3, e368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, S.; Zhang, Y.; Zhang, L.; Liu, Q.; Liu, H.X.; Hu, L.; Wei, F.R.; Steinmann, P.; Graeff-Teixeira, C.; Zhou, X.N.; et al. The complete mitochondrial genome of the rodent intra-arterial nematodes Angiostrongylus cantonensis and Angiostrongylus costaricensis. Parasitol. Res. 2012, 111, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Yong, H.S.; Song, S.L.; Eamsobhana, P.; Lim, P.E. Complete mitochondrial genome of Angiostrongylus malaysiensis lungworm and molecular phylogeny of Metastrongyloid nematodes. Acta Trop. 2016, 161, 33–40. [Google Scholar] [CrossRef]
- Yong, H.S.; Song, S.L.; Eamsobhana, P.; Goh, S.Y.; Lim, P.E. Complete mitochondrial genome reveals genetic diversity of Angiostrongylus cantonensis (Nematoda: Angiostrongylidae). Acta Trop. 2015, 152, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Gasser, R.B.; Jabbar, A.; Mohandas, N.; Schnyder, M.; Deplazes, P.; Littlewood, D.T.; Jex, A.R. Mitochondrial genome of Angiostrongylus vasorum: Comparison with congeners and implications for studying the population genetics and epidemiology of this parasite. Infect. Genet. Evol. 2012, 12, 1884–1891. [Google Scholar] [CrossRef]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alicata, J.E. The presence of Angiostrongylus cantonensis in islands of the Indian Ocean and probable role of the giant African snail, Achatina fulica, in dispersal of the parasite to the Pacific Islands. Can. J. Zool. 1966, 44, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Deshpande, O.; Roseman, C.C.; Rosenberg, N.A.; Feldman, M.W.; Cavalli-Sforza, L.L. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc. Natl. Acad. Sci. USA 2005, 102, 15942–15947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eamsobhana, P.; Song, S.L.; Yong, H.S.; Prasartvit, A.; Boonyong, S.; Tungtrongchitr, A. Cytochrome c oxidase subunit I haplotype diversity of Angiostrongylus cantonensis (Nematoda: Angiostrongylidae). Acta Trop. 2017, 171, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Bain, R.H.; Hurley, M.M. A biogeographic synthesis of the amphibians and reptiles of Indochina. Bull. Am. Mus. Nat. Hist. 2011, 360, 1–138. [Google Scholar] [CrossRef]
- Qian, M.B.; Chen, Y.D.; Liang, S.; Yang, G.J.; Zhou, X.N. The global epidemiology of clonorchiasis and its relation with cholangiocarcinoma. Infect. Dis. Poverty 2012, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- IARC. A review of human carcinogens. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; World Health Organization: Geneva, Switzerland, 2012; Volume 100B, p. 441. [Google Scholar]
- Lee, J.D.; Chung, L.Y.; Wang, L.C.; Lin, R.J.; Wang, J.J.; Tu, H.P.; Wu, Z.D.; Yen, C.M. Sequence analysis in partial genes of five isolates of Angiostrongylus cantonensis from Taiwan and biological comparison in infectivity and pathogenicity between two strains. Acta Trop. 2014, 133, 26–34. [Google Scholar] [CrossRef]
- Peng, J.; He, Z.P.; Zhang, S.; Lun, Z.R.; Wu, Z.D.; Fan, C.K.; Brown, C.L.; Cheng, P.C.; Peng, S.Y.; Yang, T.B. Phylogeography of Angiostrongylus cantonensis (Nematoda: Angiostrongylidae) in southern China and some surrounding areas. PLoS Negl. Trop. Dis. 2017, 11, e0005776. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Access Number | Location | Altitude | Longitude | References |
|---|---|---|---|---|---|
| A. cantonensis | AP017672 | Taipei, China | 25.0329 | 121.5655 | [30] |
| CL_Acan | Changle, China | 25.9313 | 119.6288 | this study | |
| DF1_Acan | Dongfang, China | 19.0535 | 108.6521 | this study | |
| DF2_Acan | Dongfang, China | 19.0535 | 108.6521 | this study | |
| GQ398121 | Lianjiang, China | 26.2052 | 119.5212 | [32] | |
| KT947978 | Thailand | [26] | |||
| MK570629 | Tenerife, Spain | 28.2916 | −16.6291 | [30] | |
| MK570630 | Hawaii, USA | 19.6310 | −156.0072 | [30] | |
| MK570631 | Mosman, Australia | −33.8293 | 151.2442 | [30] | |
| MK570632 | Fatu Hiva, French Polynesia | −17.6427 | −149.4347 | [30] | |
| NA_Acan | Nanao, China | 23.4533 | 117.0971 | this study | |
| RL_Acan | Ruili, China | 23.9477 | 97.7854 | this study | |
| SC_Acan | Shangchuan, China | 21.6613 | 112.8016 | this study | |
| SY_Acan | Sanya, China | 18.3333 | 109.4333 | this study | |
| ZX_Acan | Zixing, China | 26.0365 | 113.2463 | this study | |
| A. mackerrasae | MN793157 | Brisbane, Australia | −27.4620 | 153.0203 | [33] |
| A. malaysiensis | KT186242 | Thailand | [34] | ||
| KT947979 | Kuala Lumpur, Malaysia | 3.1201 | 101.6545 | [26] | |
| A. vasorum | JX268542 | Australia | [35] | ||
| A. costaricensis | GQ398122 | Brazil | [32] |
| MN793157 | KT186242 | KT947979 | CL_Acan | NA_Acan | GQ398121 | SY_Acan | ZX_Acan | MK570632 | MK570630 | MK570629 | AP017672 | MK570631 | KT947978 | RL_Acan | DF1_Acan | DF2_Acan | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KT186242 | 0.133 | ||||||||||||||||
| KT947979 | 0.133 | 0.002 | |||||||||||||||
| CL_Acan | 0.116 | 0.124 | 0.124 | ||||||||||||||
| NA_Acan | 0.116 | 0.125 | 0.124 | 0.001 | |||||||||||||
| GQ398121 | 0.117 | 0.125 | 0.125 | 0.002 | 0.002 | ||||||||||||
| SY_Acan | 0.115 | 0.123 | 0.123 | 0.008 | 0.007 | 0.009 | |||||||||||
| ZX_Acan | 0.116 | 0.123 | 0.123 | 0.015 | 0.015 | 0.016 | 0.014 | ||||||||||
| MK570632 | 0.118 | 0.125 | 0.125 | 0.035 | 0.035 | 0.036 | 0.034 | 0.034 | |||||||||
| MK570630 | 0.118 | 0.124 | 0.124 | 0.034 | 0.034 | 0.035 | 0.033 | 0.033 | 0.001 | ||||||||
| MK570629 | 0.118 | 0.123 | 0.123 | 0.034 | 0.034 | 0.035 | 0.033 | 0.034 | 0.011 | 0.010 | |||||||
| AP017672 | 0.118 | 0.123 | 0.123 | 0.034 | 0.034 | 0.035 | 0.033 | 0.034 | 0.011 | 0.010 | 0.001 | ||||||
| MK570631 | 0.118 | 0.123 | 0.123 | 0.034 | 0.034 | 0.035 | 0.033 | 0.033 | 0.011 | 0.010 | 0.001 | 0.001 | |||||
| KT947978 | 0.118 | 0.124 | 0.124 | 0.034 | 0.034 | 0.035 | 0.033 | 0.033 | 0.018 | 0.017 | 0.016 | 0.016 | 0.016 | ||||
| RL_Acan | 0.119 | 0.125 | 0.125 | 0.034 | 0.034 | 0.035 | 0.033 | 0.033 | 0.019 | 0.019 | 0.017 | 0.017 | 0.017 | 0.009 | |||
| DF1_Acan | 0.118 | 0.124 | 0.124 | 0.035 | 0.035 | 0.036 | 0.033 | 0.034 | 0.030 | 0.029 | 0.029 | 0.029 | 0.029 | 0.029 | 0.030 | ||
| DF2_Acan | 0.117 | 0.124 | 0.124 | 0.035 | 0.035 | 0.036 | 0.033 | 0.034 | 0.030 | 0.029 | 0.029 | 0.029 | 0.029 | 0.028 | 0.030 | 0.001 | |
| SC_Acan | 0.117 | 0.126 | 0.126 | 0.051 | 0.052 | 0.052 | 0.051 | 0.053 | 0.056 | 0.056 | 0.055 | 0.056 | 0.055 | 0.055 | 0.055 | 0.056 | 0.056 |
| Types | A. cantonensis | A. mackerrasae | A. malaysiensis | Total |
|---|---|---|---|---|
| mt genome | 8 (16 *) | 1 (1) | 1 (1) | 10 (18) |
| cox1 | 144 (426 #) | 5 (5) | 11 (23) | 160 (454) |
| cytb | 177 (655 $) | 0 (0) | 76 (76) | 253 (731) |
| nad1 | 0 (130) | 0 (0) | 0 (0) | 0 (130) |
| SSU | 13 (13) | 0 (0) | 53 (53) | 66 (66) |
| LSU | 12 (12) | 0 (0) | 53 (53) | 65 (65) |
| Total | 354 (1252) | 6 (6) | 194 (206) | 554 (1472) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, X.; Chen, S.; Duan, L.; Qian, Y.; Li, H.; Lv, S. The Global Spread Pattern of Rat Lungworm Based on Mitochondrial Genetics. Pathogens 2023, 12, 788. https://doi.org/10.3390/pathogens12060788
Tian X, Chen S, Duan L, Qian Y, Li H, Lv S. The Global Spread Pattern of Rat Lungworm Based on Mitochondrial Genetics. Pathogens. 2023; 12(6):788. https://doi.org/10.3390/pathogens12060788
Chicago/Turabian StyleTian, Xia, Shen Chen, Lei Duan, Yingjun Qian, Hongmei Li, and Shan Lv. 2023. "The Global Spread Pattern of Rat Lungworm Based on Mitochondrial Genetics" Pathogens 12, no. 6: 788. https://doi.org/10.3390/pathogens12060788