
Differentiation of African Swine Fever Virus Strains Isolated in Estonia by Multiple Genetic Markers

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Selection of Isolates
2.3. Genome Detection and Amplification of Gene Regions by Conventional PCR and Sequence Analysis
2.4. Mapping Software
3. Results
3.1. Sequencing of the IGR I73R/I329L, MGF505-5R, K145R, and O174L Gene Regions
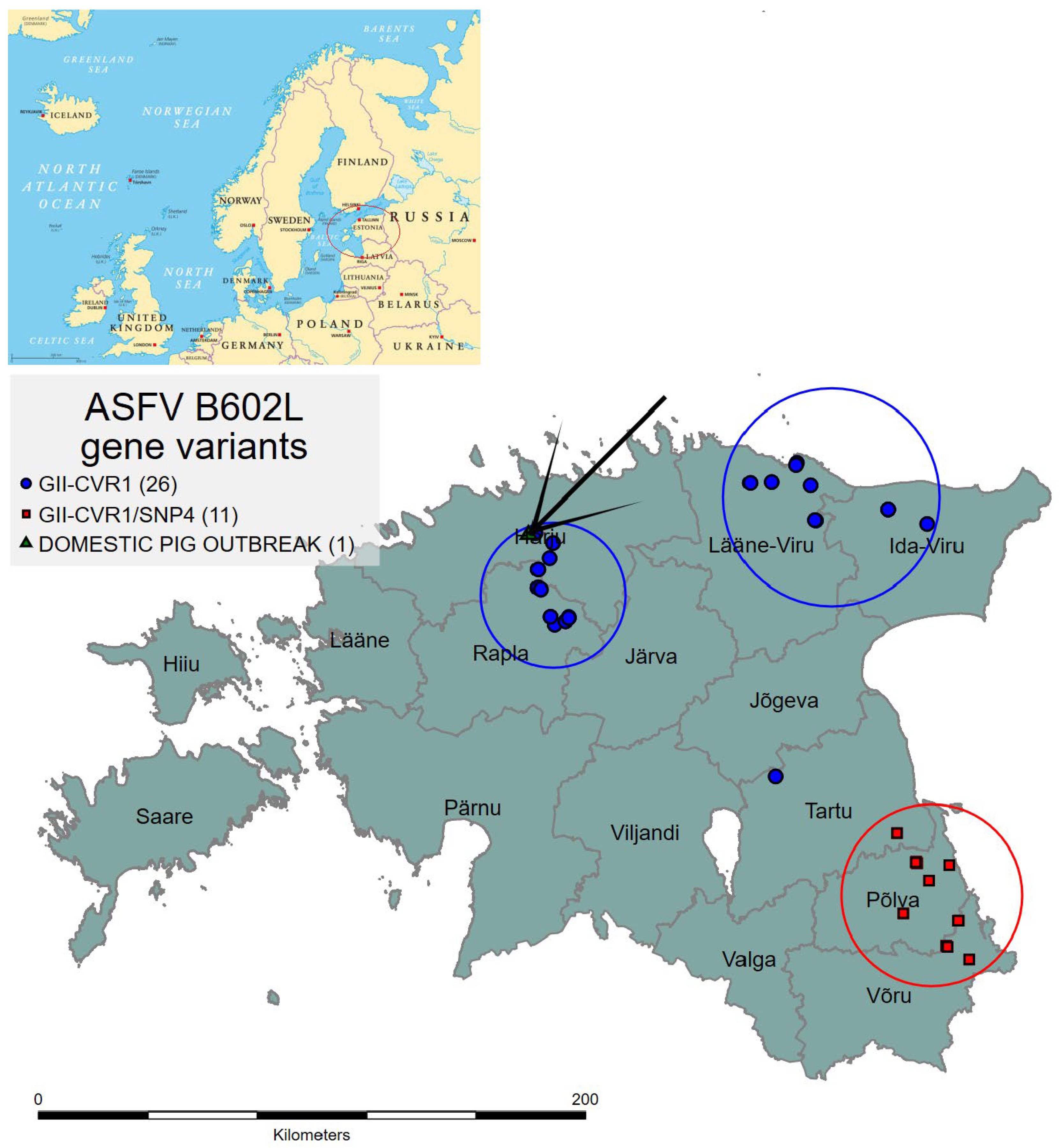
3.2. Sequencing of the B602L Gene
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alonso, C.; Borca, M.; Dixon, L.; Revilla, Y.; Rodriguez, F.; Escribano, J.M.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Asfarviridae. J. Gen. Virol. 2018, 99, 613–614. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C.; Fernández-Pinero, J.; Pelayo, V.; Gazaev, I.; Markowska-Daniel, I.; Pridotkas, G.; Nieto, R.; Fernández-Pacheco, P.; Bokhan, S.; Nevolko, O.; et al. Genetic Variation among African Swine Fever Genotype II Viruses, Eastern and Central Europe. Emerg. Infect. Dis. 2014, 20, 1544–1547. [Google Scholar] [CrossRef] [PubMed]
- Mulumba-Mfumu, L.K.; Saegerman, C.; Dixon, L.K.; Madimba, K.C.; Kazadi, E.; Mukalakata, N.T.; Oura, C.A.L.; Chenais, E.; Masembe, C.; Ståhl, K.; et al. African Swine Fever: Update on Eastern, Central and Southern Africa. Transbound. Emerg. Dis. 2019, 66, 1462–1480. [Google Scholar] [CrossRef] [PubMed]
- Mur, L. African Swine Fever (Asf)—Situation Report 4; AFRICAN SWINE FEVER 6; WOAH: Paris, France, 2022. [Google Scholar]
- Gallardo, C.; Casado, N.; Soler, A.; Djadjovski, I.; Krivko, L.; Madueño, E.; Nieto, R.; Perez, C.; Simon, A.; Ivanova, E.; et al. A Multi Gene-Approach Genotyping Method Identifies 24 Genetic Clusters within the Genotype II-European African Swine Fever Viruses Circulating from 2007 to 2022. Front. Vet. Sci. 2023, 10, 1112850. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Li, J.; Fan, X.; Liu, F.; Li, L.; Wang, Q.; Ren, W.; Bao, J.; Liu, C.; Wang, H.; et al. Molecular Characterization of African Swine Fever Virus, China, 2018. Emerg. Infect. Dis. 2018, 24, 2131–2133. [Google Scholar] [CrossRef]
- Vilem, A.; Nurmoja, I.; Niine, T.; Riit, T.; Nieto, R.; Viltrop, A.; Gallardo, C. Molecular Characterization of African Swine Fever Virus Isolates in Estonia in 2014–2019. Pathogens 2020, 9, 582. [Google Scholar] [CrossRef]
- Nurmoja, I.; Schulz, K.; Staubach, C.; Sauter-Louis, C.; Depner, K.; Conraths, F.J.; Viltrop, A. Development of African Swine Fever Epidemic among Wild Boar in Estonia—Two Different Areas in the Epidemiological Focus. Sci. Rep. 2017, 7, 12562. [Google Scholar] [CrossRef]
- Nurmoja, I.; Petrov, A.; Breidenstein, C.; Zani, L.; Forth, J.H.; Beer, M.; Kristian, M.; Viltrop, A.; Blome, S. Biological Characterization of African Swine Fever Virus Genotype II Strains from North-Eastern Estonia in European Wild Boar. Transbound. Emerg. Dis. 2017, 64, 2034–2041. [Google Scholar] [CrossRef]
- Schulz, K.; Oļševskis, E.; Viltrop, A.; Masiulis, M.; Staubach, C.; Nurmoja, I.; Lamberga, K.; Seržants, M.; Malakauskas, A.; Conraths, F.J.; et al. Eight Years of African Swine Fever in the Baltic States: Epidemiological Reflections. Pathogens 2022, 11, 711. [Google Scholar] [CrossRef]
- Nurmoja, I.; Mõtus, K.; Kristian, M.; Niine, T.; Schulz, K.; Depner, K.; Viltrop, A. Epidemiological Analysis of the 2015–2017 African Swine Fever Outbreaks in Estonia. Prev. Vet. Med. 2018, 181, 104556. [Google Scholar] [CrossRef]
- Schulz, K.; Schulz, J.; Staubach, C.; Blome, S.; Nurmoja, I.; Conraths, F.J.; Sauter-Louis, C.; Viltrop, A. African Swine Fever Re-Emerging in Estonia: The Role of Seropositive Wild Boar from an Epidemiological Perspective. Viruses 2021, 13, 2121. [Google Scholar] [CrossRef]
- Chapman, D.A.G.; Darby, A.C.; Da Silva, M.; Upton, C.; Radford, A.D.; Dixon, L.K. Genomic Analysis of Highly Virulent Georgia 2007/1 Isolate of African Swine Fever Virus. Emerg. Infect. Dis. 2011, 17, 599–605. [Google Scholar] [CrossRef]
- Zani, L.; Forth, J.H.; Forth, L.; Nurmoja, I.; Leidenberger, S.; Henke, J.; Carlson, J.; Breidenstein, C.; Viltrop, A.; Höper, D.; et al. Deletion at the 5’-End of Estonian ASFV Strains Associated with an Attenuated Phenotype. Sci. Rep. 2018, 8, 6510. [Google Scholar] [CrossRef]
- Mazur-Panasiuk, N.; Walczak, M.; Juszkiewicz, M.; Woźniakowski, G. The Spillover of African Swine Fever in Western Poland Revealed Its Estimated Origin on the Basis of O174L, K145R, MGF 505-5R and IGR I73R/I329L Genomic Sequences. Viruses 2020, 12, 1094. [Google Scholar] [CrossRef]
- Sauter-Louis, C.; Forth, J.H.; Probst, C.; Staubach, C.; Hlinak, A.; Rudovsky, A.; Holland, D.; Schlieben, P.; Göldner, M.; Schatz, J.; et al. Joining the Club: First Detection of African Swine Fever in Wild Boar in Germany. Transbound. Emerg. Dis. 2021, 68, 1744–1752. [Google Scholar] [CrossRef]
- Sauter-Louis, C.; Schulz, K.; Richter, M.; Staubach, C.; Mettenleiter, T.C.; Conraths, F.J. African Swine Fever: Why the Situation in Germany Is Not Comparable to That in the Czech Republic or Belgium. Transbound. Emerg. Dis. 2022, 69, 2201–2208. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Mazloum, A.; Van Schalkwyk, A.; Chernyshev, R.; Shotin, A.; Korennoy, F.I.; Igolkin, A.; Sprygin, A. Genetic Characterization of the Central Variable Region in African Swine Fever Virus Isolates in the Russian Federation from 2013 to 2017. Pathogens 2022, 11, 919. [Google Scholar] [CrossRef]
- Gallardo, C.; Soler, A.; Nieto, R.; Cano, C.; Pelayo, V.; Sánchez, M.A.; Pridotkas, G.; Fernandez-Pinero, J.; Briones, V.; Arias, M. Experimental Infection of Domestic Pigs with African Swine Fever Virus Lithuania 2014 Genotype II Field Isolate. Transbound. Emerg. Dis. 2017, 64, 300–304. [Google Scholar] [CrossRef]
- Administrative and Settlement Division” Counties Map. Available online: https://geoportaal.maaamet.ee/docs/haldus_asustus/maakond_shp.zip?t=20230301005655 (accessed on 15 March 2023).
- Credit Peter Hermes Furian. Available online: https://www.istockphoto.com/collaboration/boards/IlHi2WsN7ESFV9sVvQ4TGg (accessed on 25 April 2023).
- Malogolovkin, A.; Kolbasov, D. Genetic and Antigenic Diversity of African Swine Fever Virus. Virus Res. 2019, 271, 197673. [Google Scholar] [CrossRef]
- Qu, H.; Ge, S.; Zhang, Y.; Wu, X.; Wang, Z. A Systematic Review of Genotypes and Serogroups of African Swine Fever Virus. Virus Genes 2022, 58, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Mazloum, A.; van Schalkwyk, A.; Shotin, A.; Zinyakov, N.; Igolkin, A.; Chernishev, R.; Debeljak, Z.; Korennoy, F.; Sprygin, A.V. Whole-Genome Sequencing of African Swine Fever Virus from Wild Boars in the Kaliningrad Region Reveals Unique and Distinguishing Genomic Mutations. Front. Vet. Sci. 2023, 9, 1019808. [Google Scholar] [CrossRef] [PubMed]
- Mazur-Panasiuk, N.; Woźniakowski, G. The Unique Genetic Variation within the O174L Gene of Polish Strains of African Swine Fever Virus Facilitates Tracking Virus Origin. Arch. Virol. 2019, 164, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Mazloum, A.; van Schalkwyk, A.; Chernyshev, R.; Igolkin, A.; Heath, L.; Sprygin, A. A Guide to Molecular Characterization of Genotype II African Swine Fever Virus: Essential and Alternative Genome Markers. Microorganisms 2023, 11, 642. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C.; Nurmoja, I.; Soler, A.; Delicado, V.; Simón, A.; Martin, E.M.; Perez, C.P.; Nieto, R.; Arias, M. Evolution in Europe of African Swine Fever Genotype II Viruses from Highly to Moderately Virulent. Vet. Microbiol. 2018, 219, 70–79. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| County | No. of Isolates Selected | No. of Successfully Sequenced Isolates by Genome Region | No. of Isolates Selected for B602L * | |||||
|---|---|---|---|---|---|---|---|---|
| IGR I73-I329L | MGF505-5R | K145R | O174L | CVR B602L | ||||
| Harju ** | 18 | 16 | 14 | 17 | 14 | 5 | 5 | |
| Ida-Viru | 9 | 6 | 6 | 9 | 5 | 4 | 4 | |
| Jõgeva | 5 | 5 | 5 | 5 | 5 | |||
| Järva | 4 | 3 | 3 | 4 | 2 | |||
| Lääne | 7 | 6 | 6 | 6 | 5 | |||
| Lääne-Viru | 19 | 18 | 13 | 15 | 14 | 7 | 9 | |
| Põlva | 10 | 10 | 10 | 10 | 10 | 6 | 6 | |
| Pärnu | 8 | 7 | 7 | 7 | 6 | |||
| Rapla | 21 | 18 | 17 | 20 | 17 | 10 | 11 | |
| Saare | 9 | 9 | 8 | 9 | 7 | |||
| Tartu | 11 | 8 | 11 | 11 | 9 | 2 | 2 | |
| Valga | 6 | 6 | 6 | 6 | 6 | |||
| Viljandi | 7 | 7 | 6 | 7 | 6 | |||
| Võru | 12 | 11 | 11 | 11 | 9 | 4 | 5 | |
| Total | n | 146 | 130 | 123 | 137 | 115 | 38 | 42 |
| % | X | 89.0 | 84.2 | 93.8 | 78.8 | 26.0 | X | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vilem, A.; Nurmoja, I.; Tummeleht, L.; Viltrop, A. Differentiation of African Swine Fever Virus Strains Isolated in Estonia by Multiple Genetic Markers. Pathogens 2023, 12, 720. https://doi.org/10.3390/pathogens12050720
Vilem A, Nurmoja I, Tummeleht L, Viltrop A. Differentiation of African Swine Fever Virus Strains Isolated in Estonia by Multiple Genetic Markers. Pathogens. 2023; 12(5):720. https://doi.org/10.3390/pathogens12050720
Chicago/Turabian StyleVilem, Annika, Imbi Nurmoja, Lea Tummeleht, and Arvo Viltrop. 2023. "Differentiation of African Swine Fever Virus Strains Isolated in Estonia by Multiple Genetic Markers" Pathogens 12, no. 5: 720. https://doi.org/10.3390/pathogens12050720