

The Evolution of Post-Vaccine G8P[4] Group a Rotavirus Strains in Rwanda; Notable Variance at the Neutralization Epitope Sites
 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Double-Stranded RNA Extraction and cDNA Synthesis
2.3. Preparation of DNA Library for Whole Genome Sequencing
2.4. Genome Assembly
2.5. Genome Genotyping and GenBanK Accession Numbers
2.6. Sequence and Phylogenetic Analyses
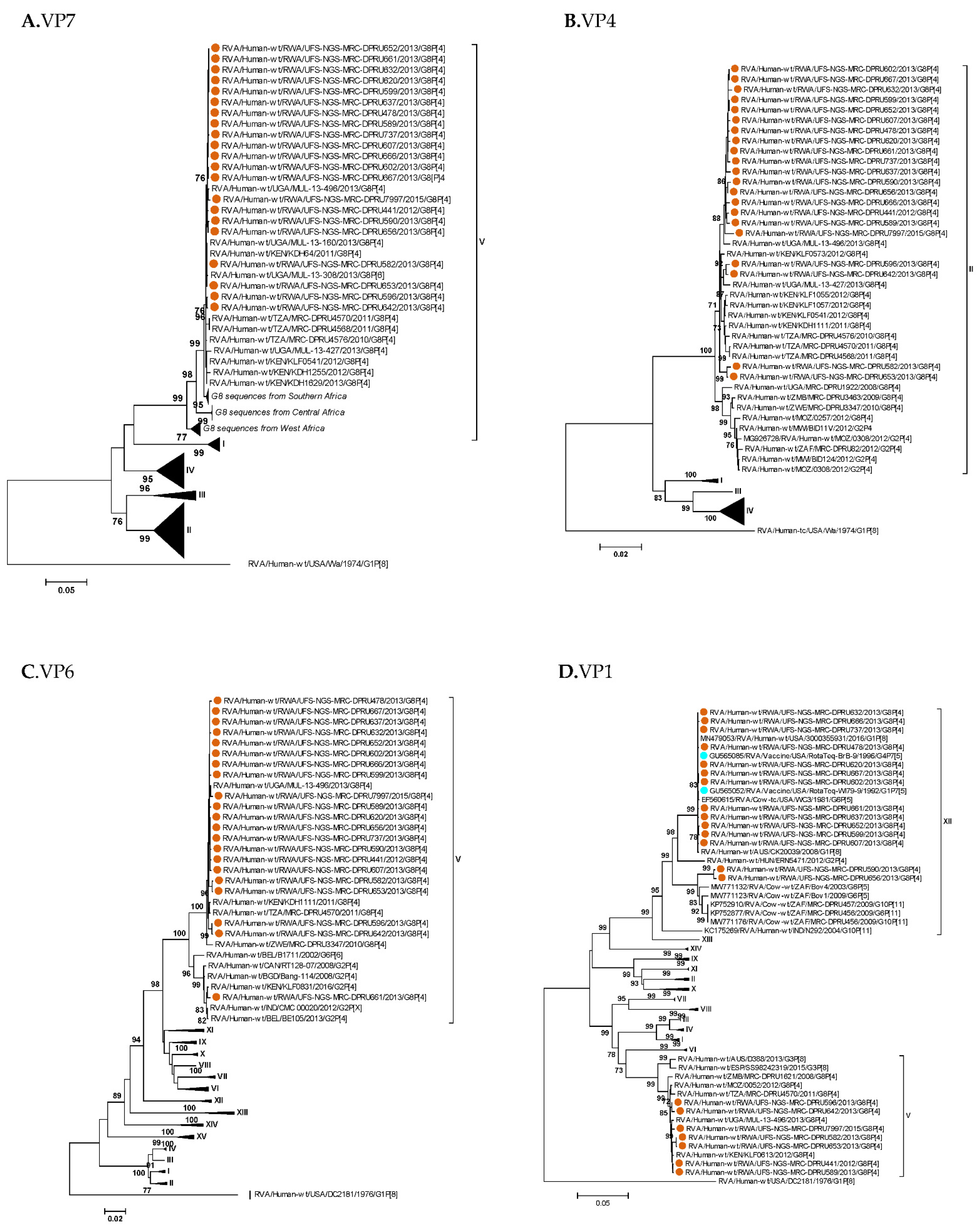
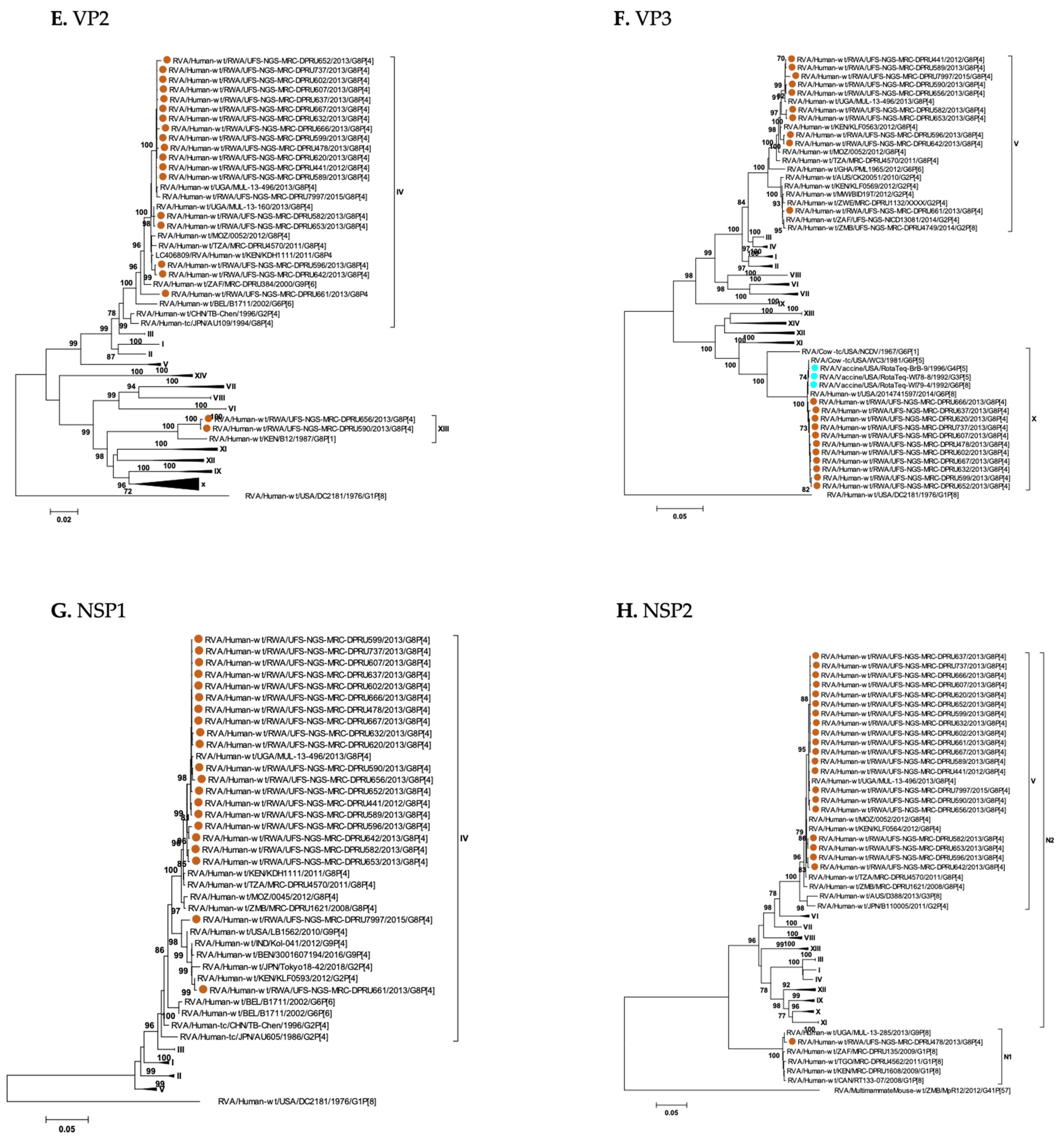
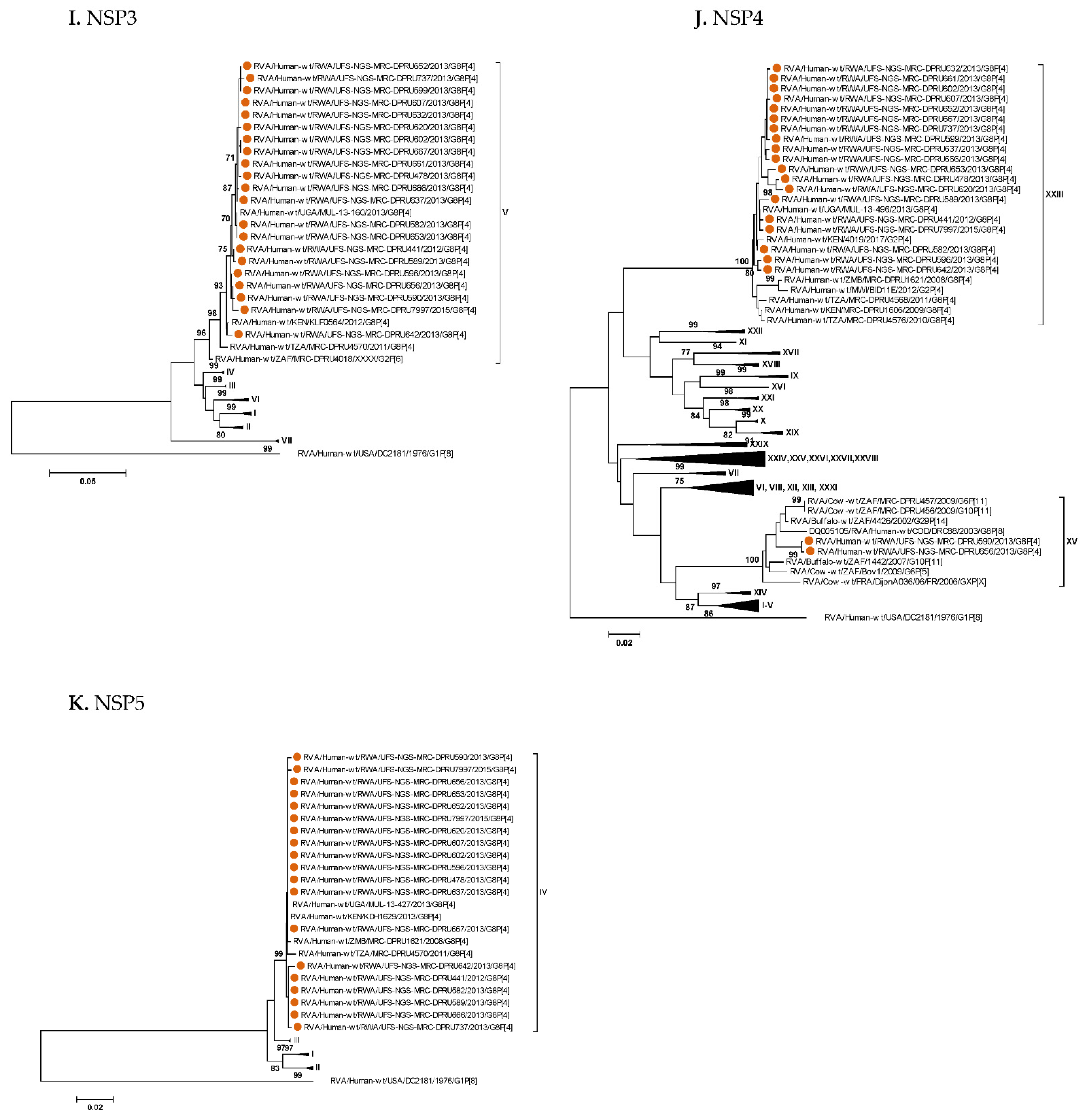
3. Results
3.1. Full Genotyping Results

{kind=link}
{kind=link}
{kind=link}
| Rwandan G8P[4] Strains | Genotype Constellations | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| VP7 | VP4 | VP6 | VP1 | VP2 | VP3 | NSP1 | NSP2 | NSP3 | NSP4 | NSP5 | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU441/2012/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (V) | C2 (IV) | M2 (V) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU478/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (XII) | C2 (IV) | M2 (X) | A2 (IV) | N1 | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU582/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (V) | C2 (IV) | M2 (V) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU589/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (V) | C2 (IV) | M2 (V) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU590/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (XII) | C2 (XIII) | M2 (V) | A2 (IV) | N2 (V) | T2 (V) | E2 (XV) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU596/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V | R2 (V) | C2 (IV) | M2 (V) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU599/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (XII) | C2 (IV) | M2 (X) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU602/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (XII) | C2 (IV) | M2 (X) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU607/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (XII) | C2 (IV) | M2 (X) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU620/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (XII) | C2 (IV) | M2 (X) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU632/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (XII) | C2 (IV) | M2 (X) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU637/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (XII) | C2 (IV) | M2 (X) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU642/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (V) | C2 (IV) | M2 (V) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU652/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (XII) | C2 (IV) | M2 (X) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU653/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V | R2 (V) | C2 (IV) | M2 (V) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU656/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (XII | C2 (XIII) | M2 (V) | A2 (IV) | N2 (V) | T2 (V) | E2 (XV) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU661/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (XII) | C2 (IV) | M2 (V) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU666/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (XII) | C2 (IV) | M2 (X) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU667/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (XII) | C2 (IV) | M2 (X) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU737/2013/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (XII) | C2 (IV) | M2 (X) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU7997/2015/G8P[4] | G8 (V) | P[4] (II) | I2 (V) | R2 (V) | C2 (IV) | M2 (V) | A2 (IV) | N2 (V) | T2 (V) | E2 (XXIII) | H2 (IV) |



3.2. Analysis of Neutralization Epitopes
3.3. Phylogenetic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cohen, A.L.; Platts-Mills, J.A.; Nakamura, T.; Operario, D.J.; Antoni, S.; Mwenda, J.M.; Weldegebriel, G.; Rey-Benito, G.; De Oliveira, L.H.; Ortiz, C.; et al. Aetiology and incidence of diarrhoea requiring hospitalisation in children under 5 years of age in 28 low-income and middle-income countries: Findings from the Global Pediatric Diarrhea Surveillance network. BMJ Glob. Health 2022, 7, e009548. [Google Scholar] [CrossRef]
- Sibomana, H.; Rugambwa, C.; Sayinzoga, F.; Iraguha, G.; Uwimana, J. Impact of routine rotavirus vaccination on all-cause and rotavirus hospitalizations during the first four years following vaccine introduction in Rwanda. Vaccine 2018, 36, 7135–7141. [Google Scholar] [CrossRef] [PubMed]
- Burnett, E.; Parashar, U.D.; Tate, J.E. Real-world efffectiveness of rotavirus vaccines 2006–2019: A literature review and meta-analysis. Lancent Glob. Health 2020, 8, 1195–1202. [Google Scholar] [CrossRef]
- Estes, M.K.; Greenberg, H.B. Rotaviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Heath/Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2013; pp. 1347–1401. [Google Scholar]
- Matthijnssens, J.; Ciarlet, M.; Rahman, M.; Attoui, H.; Bányai, K.; Estes, M.K.; Gentsch, J.R.; Iturriza-Gómara, M.; Kirkwood, C.D.; Martella, V.; et al. Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch. Virol. 2008, 153, 1621–1629. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Ciarlet, M.; McDonald, S.M.; Attoui, H.; Bányai, K.; Brister, J.R.; Buesa, J.; Esona, M.D.; Estes, M.K.; Gentsch, J.R.; et al. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch. Virol. 2011, 156, 1397–1413. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Van Ranst, M. Genotype constellation and evolution of group A rotaviruses infecting humans. Curr. Opin Virol. 2012, 2, 426–433. [Google Scholar] [CrossRef]
- Mhango, C.; Banda, A.; Chinyama, E.; Mandolo, J.J.; Kumwenda, O.; Malamba-Banda, C.; Barnes, K.G.; Kumwenda, B.; Jambo, K.; Donato, C.M.; et al. Comparative whole genome analysis reveals re-emergence of typical human Wa-like and DS-1-like G3 rotaviruses after Rotarix vaccine introduction in Malawi. medRxiv 2022. [Google Scholar] [CrossRef]
- Mwanga, M.J.; Verani, J.R.; Omore, R.; Tate, J.E.; Parashar, U.D.; Murunga, N.; Gicheru, E.; Breiman, R.F.; Nokes, D.J.; Agoti, C.N. Multiple introductions and predominance of rotavirus group A genotype G3P[8] in coastal Kenya in 2018, 4 years after nationwide vaccine introduction. Pathogens 2020, 9, 981. [Google Scholar] [CrossRef]
- Fukuda, S.; Tacharoenmuang, R.; Guntapong, R.; Upachai, S.; Singchai, P.; Ide, T.; Hatazawa, R.; Sutthiwarakom, K.; Kongjorn, S.; Onvimala, N.; et al. Full genome characterization of novel DS-1-like G9P[8] rotavirus strains that have emerged in Thailand. PLoS ONE 2020, 15, e0231099. [Google Scholar] [CrossRef]
- Jere, K.C.; Chaguza, C.; Bar-Zeev, N.; Lowe, J.; Peno, C.; Kumwenda, B.; Nakagomi, O.; Tate, J.E.; Parashar, U.D.; Heyderman, R.S.; et al. Emergence of double-and triple-gene reassortant G1P[8] rotaviruses possessing a DS-1-like backbone after rotavirus vaccine introduction in Malawi. J. Virol. 2018, 92, e01246-17. [Google Scholar] [CrossRef]
- Maringa, W.M.; Simwaka, J.; Mwangi, P.N.; Mpabalwani, E.M.; Mwenda, J.M.; Mphahlele, M.J.; Seheri, M.L.; Nyaga, M.M. Whole Genome Analysis of Human Rotaviruses Reveals Single Gene Reassortant Rotavirus Strains in Zambia. Viruses 2021, 13, 1872. [Google Scholar] [CrossRef]
- Wandera, E.A.; Hatazawa, R.; Tsutsui, N.; Kurokawa, N.; Kathiiko, C.; Mumo, M.; Waithira, E.; Wachira, M.; Mwaura, B.; Nyangao, J.; et al. Genomic characterization of an African G4P[6] human rotavirus strain identified in a diarrheic child in Kenya: Evidence for porcine-to-human interspecies transmission and reassortment. Infect. Genet. Evol. 2021, 96, 105133. [Google Scholar] [CrossRef]
- Maringa, W.M.; Mwangi, P.N.; Simwaka, J.; Mpabalwani, E.M.; Mwenda, J.M.; Peenze, I.; Esona, M.D.; Mphahlele, M.J.; Seheri, M.L.; Nyaga, M.M. Molecular characterisation of a rare reassortant porcine-like G5P[6] rotavirus strain detected in an unvaccinated child in Kasama, Zambia. Pathogens 2020, 9, 663. [Google Scholar] [CrossRef] [PubMed]
- Rasebotsa, S.; Mwangi, P.N.; Mogotsi, M.T.; Sabiu, S.; Magagula, N.B.; Rakau, K.; Uwimana, J.; Mutesa, L.; Muganga, N.; Murenzi, D.; et al. Whole genome and in-silico analyses of G1P[8] rotavirus strains from pre-and post-vaccination periods in Rwanda. Sci. Rep. 2020, 10, 13460. [Google Scholar] [CrossRef]
- Midgley, S.E.; Banyai, K.; Buesa, J.; Halaihel, N.; Hjulsager, C.K. Diversity and zoonotic potential of rotaviruses in swine and cattle across Europe. Vet. Microbiol. 2012, 156, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Alkan, F.; Ozkul, A.; Oguzoglu, T.C.; Timurkan, M.O.; Caliskan, E. Distribution of G (VP7) and P (VP4) genotypes of group A bovine rotaviruses from Turkish calves with diarrhea, 1997–2008. Vet. Microbiol. 2010, 141, 231–237. [Google Scholar] [CrossRef]
- Palombo, E.A. Genetic analysis of Group A rotaviruses: Evidence for interspecies transmission of rotavirus genes. Virus Genes 2002, 24, 11–20. [Google Scholar] [CrossRef]
- Monini, M.; Cappuccini, F.; Battista, P.; Falcone, E.; Lavazza, A. Molecular characterization of bovine rotavirus strains circulating in northern Italy, 2003–2005. Vet. Microbiol. 2008, 129, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Fodha, I.; Boumaiza, A.; Chouikha, A.; Dewar, J.; Armah, G. Detection of group A rotavirus strains circulating in calves in Tunisia. J. Vet. Med. B Infect. Dis. Vet. Public Health 2005, 52, 49–50. [Google Scholar] [CrossRef]
- Nyaga, M.M.; Stucker, K.M.; Esona, M.D.; Jere, K.C.; Mwinyi, B.; Shonhai, A.; Tsolenyanu, E.; Mulindwa, A.; Chibumbya, J.N.; Adolfine, H.; et al. Whole-genome analyses of DS-1-like human G2P [4] and G8P[4] rotavirus strains from Eastern, Western and Southern Africa. Virus Genes 2014, 49, 196–207. [Google Scholar] [CrossRef]
- Nakagomi, T.; Doan, Y.H.; Dove, W.; Ngwira, B.; Itturiza-Gomara, M. G8 rotaviruses with conserved genotype constellations detected in Malawi over 10 years (1997–2007) display frequent gene reassortment among strains co- circulating in humans. J. Gen. Virol. 2013, 94, 1273–1295. [Google Scholar] [CrossRef]
- Mwenda, J.M.; Ntoto, K.M.; Abebe, A.; Enweronu-Laryea, C.; Amina, I. Burden and epidemiology of rotavirus diarrhea in selected African countries: Preliminary results from the African Rotavirus Surveillance Network. J. Infect. Dis. 2010, 202, 5–11. [Google Scholar] [CrossRef]
- Todd, S.; Page, N.A.; Steele, A.D.; Peenze, I.; Cunliffe, N.A. Rotavirus strain types circulating in Africa: Review of studies published during 1997–2006. J. Infect. Dis. 2010, 202, 34–42. [Google Scholar] [CrossRef]
- Page, N.; Esona, M.; Seheri, M.; Nyangao, J.; Bos, P. Characterization of genotype G8 strains from Malawi, Kenya, and South Africa. J. Med. Virol. 2010, 82, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Nielson, N.M.; Eugen-Olson, J.; Aaby, P.; Molbak, K.; Rodrigues, A. Characterisation of rotavirus strains among hospitalized and nonhospitalized children in Guinea-Bissau, 2002: A high frequency of mixed infections with serotype G8. J. Clin. Virol. 2005, 34, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Jere, K.C.; Mlera, L.; O’Neill, H.G.; Peenze, I.; van Dijk, A.A. Whole genome sequence analysis of three African bovine rotaviruses reveal that theyemerged through multiple reassortment events between rotaviruses from different mammalian species. Vet. Microbiol. 2012, 159, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Esona, M.D.; Geyer, A.; Page, N.; Trabelsi, A.; Fodha, I.; Aminu, M.; Agbaya, V.; Tsion, B.; Kerin, T.; Armah, G.; et al. Genomic characterization of human rotavirus G8 strains from the African rotavirus network: Relationship to animal rotaviruses. J. Med. Virol. 2009, 81, 937–951. [Google Scholar] [CrossRef]
- Cunliffe, N.A.; Gentsch, J.R.; Kirkwood, C.D.; Gondwe, J.S.; Dove, W.; Nakagomi, O.; Nakagomi, T.; Hoshino, Y.; Bresee, J.S.; Glass, R.I.; et al. Molecular and serologic characterization of novel serotype G8 human rotavirus strains detected in Blantyre, Malawi. Virology 2000, 274, 309–320. [Google Scholar] [CrossRef]
- Steele, A.D.; Neuzil, K.M.; Cunliffe, N.A.; Madhi, S.A.; Bos, P.; Ngwira, B.; Witte, D.; Todd, S.; Louw, C.; Kirsten, M.; et al. Human rotavirus vaccine Rotarix™ provides protection against diverse circulating rotavirus strains in African infants: A randomized controlled trial. BMC Infect. Dis. 2012, 12, 213. [Google Scholar] [CrossRef]
- Justino, M.C.; Linhares, A.C.; Lanzieri, T.M.; Miranda, Y.; Mascarenhas, J.D.A.P.; Abreu, E.; Guerra, S.F.S.; Oliveira, A.S.L.; da Silva, V.B.; Sanchez, N.; et al. Effectiveness of the monovalent G1P[8] human rotavirus vaccine against hospitalization for severe G2P[4] rotavirus gastroenteritis in Belem, Brazil. Pediatr. Infect. Dis. J. 2011, 30, 396–401. [Google Scholar] [CrossRef]
- Correia, J.B.; Patel, M.M.; Nakagomi, O.; Montenegro, F.M.U.; Germano, E.M.; Correia, N.B.; Cuevas, L.E.; Parashar, U.D.; Cunliffe, N.; Nakagomi, T. Effectiveness of monovalent rotavirus vaccine (Rotarix) against severe diarrhea caused by serotypically unrelated G2P[4] strains in Brazil. J. Infect. Dis. 2010, 201, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Madhi, S.A.; Cunliffe, N.A.; Steele, A.D.; Witte, D.; Kirsten, M.; Louw, C.; Ngwira, B.; Victor, J.C.; Gillard, P.H.; Cheuvart, B.B.; et al. Effect of human rotavirus vaccine on severe diarrhea in African infants. N. Engl. J. Med. 2010, 362, 289–298. [Google Scholar] [CrossRef]
- De Vos, B.; Han, H.H.; Bouckenooghe, A.; Debrus, S.; Gillard, P.; Ward, R.; Cheuvart, B. Live attenuated human rotavirus vaccine, RIX4414, provides clinical protection in infants against rotavirus strains with and without shared G and P genotypes: Integrated analysis of randomized controlled trials. Pediatr. Infect. Dis. J. 2009, 28, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Pitzer, V.E.; Viboud, C.; Simonsen, L.; Steiner, C.; Panozzo, C.A.; Alonso, W.J.; Miller, M.A.; Glass, R.I.; Glasser, J.W.; Parashar, U.D.; et al. Demographic variability, vaccination, and the spatiotemporal dynamics of rotavirus epidemics. Science 2009, 325, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Atchison, C.; Lopman, B.; Edmunds, W.J. Modelling the seasonality of rotavirus disease and the impact of vaccination in England and Wales. Vaccine 2010, 28, 3118–3126. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Nakagomi, T.; Naghipour, M.; Nakagomi, O. Modeling seasonal variation in rotavirus hospitalizations for use in evaluating the effect of rotavirus vaccine. J. Med. Virol. 2010, 82, 1468–1474. [Google Scholar] [CrossRef]
- Andrews, S.F. A Quality Control Tool for High Throughput Sequence Data [Online]. 2010. Available online: https”//www.bioinformatics.babraham.ac.uk/projects/fastqc) (accessed on 22 December 2022).
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Sayers, E.W.; Agarwala, R.; Bolton, E.E.; Brister, J.R.; Canese, K.; Clark, K.; Connor, R.; Fiorini, N.; Funk, K.; Hefferon, T.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2019, 47, D23. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schäffer, A.A.; Brister, J.R. Virus Variation Resource–improved response to emergent viral outbreaks. Nucleic Acids Res. 2017, 45, D482–D490. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.T.; Settembre, E.C.; Trask, S.D.; Greenberg, H.B.; Harrison, S.C.; Dormitzer, P.R. Structure of rotavirus outer-layer protein VP7 bound with a neutralizing Fab. Science 2009, 324, 1444–1447. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.J.; Russell, R.B. Amino acid properties and consequences of substitutions. Bioinform. Genet. 2003, 317, 289. [Google Scholar]
- Dormitzer, P.R.; Sun, Z.Y.J.; Wagner, G.; Harrison, S.C. The rhesus rotavirus VP4 sialic acid binding domain has a galectin fold with a novel carbohydrate binding site. EMBO J. 2002, 21, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Agbemabiese, C.A.; Nakagomi, T.; Doan, Y.H.; Nakagomi, O. Whole genomic constellation of the first human G8 rotavirus strain detected in Japan. Infect. Genet. Evol. 2015, 35, 184–193. [Google Scholar] [CrossRef]
- Agbemabiese, C.A.; Nakagomi, T.; Doan, Y.H.; Do, L.P.; Damanka, S.; Armah, G.E.; Nakagomi, O. Genomic constellation and evolution of Ghanaian G2P [4] rotavirus strains from a global perspective. Infect. Genet. Evol. 2016, 45, 122–131. [Google Scholar] [CrossRef]
- Agbemabiese, C.A.; Nakagomi, T.; Damanka, S.A.; Dennis, F.E.; Lartey, B.L. Sub-genotype phylogeny of the non-G, non-P genes of genotype 2 Rotavirus A strains. PLoS ONE 2019, 14, e0217422. [Google Scholar] [CrossRef]
- Ianiro, G.; Delogu, R.; Bonomo, P.; Castiglia, P.; Ruggeri, F.M.; Fiore, L. Molecular characterization of human G8P[4] rotavirus strains in Italy: Proposal of a more complete subclassification of the G8 genotype in three major lineages. Infect. Genet. Evol. 2014, 21, 129–133. [Google Scholar] [CrossRef]
- Pietsch, C.; Petersen, L.; Patzer, L.; Liebert, U.G. Molecular characteristics of German G8P[4] rotavirus strain GER1H-09 suggest that a genotyping and subclassification update is required for G8. J. Clin. Microbiol. 2009, 47, 3569–3576. [Google Scholar] [CrossRef]
- McDonald, S.M.; Matthijnssens, J.; McAllen, J.K.; Hine, E.; Overton, L.; Wang, S.; Lemey, P.; Zeller, M.; Van Ranst, M.; Spiro, D.J.; et al. Evolutionary dynamics of human rotaviruses: Balancing reassortment with preferred genome constellations. PLoS Pathog. 2009, 5, e1000634. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Mullick, S.; Deb, A.K.; Panda, S.; Chawla-Sarkar, M. First report of human rotavirus G8P[4] gastroenteritis in India: Evidence of ruminants-to-human zoonotic transmission. J. Med. Virol. 2013, 85, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Rasebotsa, S.; Uwimana, J.; Mogotsi, M.T.; Rakau, K.; Magagula, N.B.; Seheri, M.L.; Mwenda, J.M.; Mphahlele, M.J.; Sabiu, S.; Mihigo, R.; et al. Whole-Genome Analyses Identifies Multiple Reassortant Rotavirus Strains in Rwanda Post-Vaccine Introduction. Viruses. 2021, 12, 95. [Google Scholar] [CrossRef]
- Mokoena, F.; Seheri, M.L.; Nyaga, M.M.; Magagula, N.; Mukaratirwa, A.; Mulindwa, A.; Abebe, A.; Boula, A.; Enyonam, T.; Rakau, K.; et al. Whole Genome Analysis of African G12P[6] and G12P[8] Rotaviruses Provides Evidence of Porcine-Human Reassortment at NSP2, NSP3, and NSP4. Front. Microbiol. 2021, 11, 604444. [Google Scholar] [CrossRef]
- Strydom, A.; Donato, C.M.; Nyaga, M.M.; Boene, S.S.; Peenze, I.; Mogotsi, M.T.; João, E.D.; Munlela, B.; Potgieter, A.C.; Seheri, M.L.; et al. Genetic Characterisation of South African and Mozambican Bovine Rotaviruses Reveals a Typical Bovine-like Artiodactyl Constellation Derived through Multiple Reassortment Events. Pathogens 2021, 12, 1308. [Google Scholar] [CrossRef]
- Katz, E.M.; Esona, M.D.; Betrapally, N.S.; Lucia, A.; Neira, Y.R.; Rey, G.J.; Bowen, M.D. Whole-gene analysis of inter-genogroup reassortant rotaviruses from the Dominican Republic: Emergence of equine-like G3 strains and evidence of their reassortment with locally-circulating strains. Virology 2009, 534, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Burnett, E.; Parashar, U.D.; Winn, A.; Tate, J.E. Trends in Rotavirus Laboratory Detections and Internet Search Volume Before and After Rotavirus Vaccine Introduction and in the Context of the Coronavirus Disease 2019 Pandemic—United States, 2000–2021. J. Infect. Dis. 2022, 226, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Heaton, P.M.; Ciarlet, M. The pentavalent rotavirus vaccine: Discovery to licensure and beyond. Clin. Infect. Dis. 2007, 45, 1618–1624. [Google Scholar] [CrossRef]
- Markkula, J.; Hemming, M.; Vesikari, T. Detection of vaccine-derived rotavirus strains in nonimmunocompromised children up to 3–6 months after RotaTeq vaccination. Pediatr. Infect. Dis. J. 2015, 34, 296–298. [Google Scholar] [CrossRef]
- Than, V.T.; Jeong, S.; Kim, W. Characterization of RotaTeq vaccine-derived rotaviruses in South Korean infants with rotavirus gastroenteritis. J. Med. Virol. 2015, 87, 112–116. [Google Scholar] [CrossRef]
- Donato, C.M.; Ch’Ng, L.S.; Boniface, K.F.; Crawford, N.W.; Buttery, J.P.; Lyon, M.; Bishop, R.F.; Kirkwood, C.D. Identification of strains of rotateq rotavirus vaccine in infants with gastroenteritis following routine vaccination. J. Infect. Dis. 2012, 206, 377–383. [Google Scholar] [CrossRef]
- Payne, D.C.; Edwards, K.M.; Bowen, M.D.; Keckley, E.; Peters, J.; Esona, M.D.; Teel, E.N.; Kent, D.; Parashar, U.D.; Gentsch, J.R. Sibling transmission of vaccine-derived rotavirus (RotaTeq) associated with rotavirus gastroenteritis. Pediatrics. 2010, 125, e438–e441. [Google Scholar] [CrossRef]
- Simsek, C.; Bloemen, M.; Jansen, D.; Descheemaeker, P.; Reynders, M.; Van Ranst, M.; Matthijnssens, J. Rotavirus vaccine-derived cases in Belgium: Evidence for reversion of attenuating mutations and alternative causes of gastroenteritis. Vaccine 2022, 40, 5114–5125. [Google Scholar] [CrossRef]
- Gower, C.M.; Dunning, J.; Nawaz, S.; Allen, D.; Ramsay, M.E.; Ladhani, S. Vaccine-derived rotavirus strains in infants in England. Arch. Dis. Child. 2020, 105, 553–557. [Google Scholar] [CrossRef]
- Kaneko, M.; Takanashi, S.; Thongprachum, A.; Hanaoka, N.; Fujimoto, T.; Nagasawa, K.; Kimura, H.; Okitsu, S.; Mizuguchi, M.; Ushijima, H. Identification of vaccine-derived rotavirus strains in children with acute gastroenteritis in Japan, 2012–2015. PLoS ONE 2017, 12, e0184067. [Google Scholar] [CrossRef]
- Kaplon, J.; Cros, G.; Ambert-Balay, K.; Leruez-Ville, M.; Chomton, M.; Fremy, C.; Pothier, P.; Blanche, S. Rotavirus vaccine virus shedding, viremia and clearance in infants with severe combined immune deficiency. Pediatr. Infect. Dis. J. 2015, 34, 326–328. [Google Scholar] [CrossRef]
- Bucardo, F.; Rippinger, C.M.; Svensson, L.; Patton, J.T. Vaccine-derived NSP2 segment in rotaviruses from vaccinated children with gastroenteritis in Nicaragua. Infect. Genet. Evol. 2012, 12, 1282–1294. [Google Scholar] [CrossRef]
- Trinh, Q.D.; Nguyen, T.A.; Phan, T.G.; Khamrin, P.; Yan, H.; Le Hoang, P.; Maneekarn, N.; Li, Y.; Yagyu, F.; Okitsu, S.; et al. Sequence analysis of the VP7 gene of human rotavirus G1 isolated in Japan, China, Thailand, and Vietnam in the context of changing distribution of rotavirus G-types. J. Med. Virol. 2007, 79, 1009–1016. [Google Scholar] [CrossRef]
- Ahmed, K.; Nakagomi, T.; Nakagomi, O. Molecular identification of a novel G1 VP7 gene carried by a human rotavirus with a super-short RNA pattern. Virus Genes 2007, 35, 141–145. [Google Scholar] [CrossRef]
- Monnier, N.; Higo-Moriguchi, K.; Sun, Z.-Y.J.; Prasad, B.V.V.; Taniguchi, K.; Dormitzer, P.R. High-resolution molecular and antigen structure of the VP8* core of a sialic acid-independent human rotavirus strain. J. Virol. 2006, 80, 1513–1523. [Google Scholar] [CrossRef]
- Kirkwood, C.D.; Boniface, K.; Barnes, G.L.; Bishop, R.F. Distribution of rotavirus genotypes after introduction of rotavirus vaccines, Rotarix® and RotaTeq®, into the National Immunization Program of Australia. Pediatr. Infect. Dis. J. 2011, 30, 48–53. [Google Scholar] [CrossRef]
| Neutralization Epitopes | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7-1a | 7-1b | 7-2 | ||||||||||||||||||||||||||||
| 87 | 91 | 94 | 96 | 97 | 98 | 99 | 100 | 104 | 123 | 125 | 129 | 130 | 291 | 201 | 211 | 212 | 213 | 238 | 242 | 143 | 145 | 146 | 147 | 148 | 190 | 217 | 221 | 264 | ||
| Vaccine strains | JN849114/RVA/Vaccine/USA/Rotarix-A41CB052A/1988/G1P[8] | T | T | N | G | E | W | K | D | Q | S | V | V | D | K | Q | N | V | D | N | T | K | D | Q | N | L | S | M | N | G |
| GU565057/RVA/Vaccine/USA/RotaTeq-WI79-9/1992/G1P[5] | T | T | N | G | D | W | K | D | Q | S | V | V | D | K | Q | N | V | D | N | T | K | D | Q | S | L | S | M | N | G | |
| GU565068/RVA/Vaccine/USA/RotaTeq-SC2-9/1992/G2P[5] | A | N | S | D | E | W | E | N | Q | D | T | M | N | K | Q | D | V | S | N | S | R | D | N | T | S | D | I | S | G | |
| GU565079/RVA/Vaccine/USA/RotaTeq-WI78-8/1992/G3P[5] | T | T | N | N | S | W | K | D | Q | D | A | V | D | K | Q | D | A | N | K | D | K | D | A | T | L | S | E | A | G | |
| GU565090/RVA/Vaccine/USA/RotaTeq-BrB-9/1996/G4P[5] | S | T | S | T | E | W | K | D | Q | N | L | I | D | K | Q | D | T | A | D | T | R | A | S | G | E | S | T | S | G | |
| GU565046/RVA/Vaccine/USA/RotaTeq-WI79-4/1992/G6P[8] | V | N | A | T | E | W | K | D | Q | D | A | V | E | K | Q | N | P | D | N | A | K | D | S | T | Q | S | T | T | G | |
| FJ361209/RVA/Vaccine/IND/Rotavac-116E/AG/G9P[11] | I | T | G | T | E | W | K | G | Q | D | A | I | D | K | Q | N | T | A | D | N | K | N | S | T | L | S | E | N | G | |
| AB045372/RVA/Vaccine/IND/Rotasill-Au32/2016/G9 | A | T | G | T | E | W | K | D | Q | D | A | I | D | K | Q | N | T | A | D | T | K | D | S | T | L | S | E | S | G | |
| Study strains | RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU441/2012/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU478/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU582/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU589/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU590/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU596/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | N | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU599/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU602/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU607/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU620/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU632/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU637/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU642/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | N | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU652/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU653/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU656/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU661/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU666/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU667/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU737/2013/G8P[4] | A | T | A | S | S | W | K | D | Q | D | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU7997/2015/G8P[4] | A | T | A | S | S | W | K | D | Q | N | A | I | N | K | Q | D | T | T | N | T | K | N | A | D | S | S | E | A | G | |
| Neutralization Epitopes | ||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8-1 | 8-2 | 8-3 | 8-4 | 5-1 | 5-2 | 5-3 | 5-4 | 5-5 | ||||||||||||||||||||||||||||||
| Strain | ||||||||||||||||||||||||||||||||||||||
| 100 | 146 | 148 | 150 | 188 | 190 | 192 | 193 | 194 | 195 | 196 | 180 | 183 | 113 | 114 | 115 | 116 | 125 | 131 | 132 | 133 | 135 | 87 | 88 | 89 | 384 | 386 | 388 | 393 | 394 | 398 | 440 | 441 | 434 | 459 | 429 | 306 | ||
| Vaccine strains | RVA/Vaccine/USA/Rotarix-A41CB052A/1988/G1P[8] | D | S | Q | E | S | T | N | L | N | N | I | T | A | N | P | V | D | S | S | N | D | N | N | T | N | Y | F | I | W | P | G | R | T | P | E | L | R |
| RVA/Vaccine/USA/RotaTeq-WI79-4/1992/G6P[8] | D | S | Q | E | S | T | N | L | N | D | I | T | A | N | P | V | D | N | R | N | D | D | N | T | N | Y | F | L | W | P | G | R | T | P | E | L | R | |
| Study strains | RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU441/2012/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU478/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU582/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU589/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU590/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU596/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU599/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU602/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU607/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU620/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU632/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU637/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU642/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU652/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU653/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU656/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU661/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU666/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU667/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU737/2013/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| RVA/Human-wt/RWA/UFS-NGS-MRC-DPRU7997/2015/G8P[4] | D | S | Q | E | S | T | D | L | N | N | I | T | A | S | Q | T | N | N | E | N | N | D | N | T | D | Y | F | L | W | P | G | R | T | P | E | L | R | |
| Genome Segment | Lineage | Closest Strain from GenBank | Nucleotide Similarity Range (%) | Country |
|---|---|---|---|---|
| VP7 | V | LC177390-RVA/Human-wt/KEN/KDH64/2011/G8P[4] | 99.7–100 | Kenya |
| VP4 | II | MZ097182-RVA/Human-wt/KEN/KLF1055/2012/G8P[4] | 98.8–100 | Kenya |
| VP6 | V | KX655466-RVA/Human-wt/UGA/MUL-13–496/2013/G8P[4] | 99.5–99.9 | Uganda |
| VP1 | V | MZ094284-RVA/Human-wt/KEN/KLF0613/2012/G8P[4] | 99.6–99.9 | Kenya |
| XII | RVA/Vaccine/USA/RotaTeq-BrB-9/1996/G4P[5] | 99.9–100 | USA | |
| XII | RVA/Cow-wt/ZAF/Bov1/2009/G6P[5] | 97.5–97.6 | South Africa | |
| VP2 | IV | KX655463-RVA/Human-wt/UGA/MUL-13-496/2013/G8P[4] | 99.5–99.9 | Uganda |
| XIII | HM627543-RVA/Human-wt/KEN/B12/1987/G8P[1] | 96.3–96.4 | Kenya | |
| VP3 | V | KX655464-RVA/Human-wt/UGA/MUL-13-496/2013/G8P[4] | 99.3–99.9 | Uganda |
| X | GU565043-RVA/Vaccine/USA/RotaTeq-WI79-4/1992/G6P[8] | 99.6–99.9 | USA | |
| NSP1 | IV | KX655468-RVA/Human-wt/UGA/MUL-13-496/2013/G8P[4] | 99.3–99.9 | Uganda |
| NSP2 | V | KX655469-RVA/Human-wt/UGA/MUL-13-496/2013/G8P[4] | 99.6–100 | Uganda |
| NSP3 | V | KX655503-RVA/Human-wt/UGA/MUL-13-160/2013/G8P[4]; | 99.4–100 | Uganda |
| NSP4 | XV | KX655471-RVA/Human-wt/UGA/MUL-13-496/2013/G8P[4] | 99.2–100 | Uganda |
| XXIII | MT234349- RVA/Buffalo-wt/ZAF/1442/2007/G10P[11] | 97.5–97.7 | South Africa | |
| NSP5 | IV | LC406840- RVA/Human-wt/KEN/KDH1629/2013/G8P[4] | 99.5–100 | Kenya |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mwangi, P.N.; Potgieter, R.-L.; Uwimana, J.; Mutesa, L.; Muganga, N.; Murenzi, D.; Tusiyenge, L.; Mwenda, J.M.; Mogotsi, M.T.; Rakau, K.; et al. The Evolution of Post-Vaccine G8P[4] Group a Rotavirus Strains in Rwanda; Notable Variance at the Neutralization Epitope Sites. Pathogens 2023, 12, 658. https://doi.org/10.3390/pathogens12050658
Mwangi PN, Potgieter R-L, Uwimana J, Mutesa L, Muganga N, Murenzi D, Tusiyenge L, Mwenda JM, Mogotsi MT, Rakau K, et al. The Evolution of Post-Vaccine G8P[4] Group a Rotavirus Strains in Rwanda; Notable Variance at the Neutralization Epitope Sites. Pathogens. 2023; 12(5):658. https://doi.org/10.3390/pathogens12050658
Chicago/Turabian StyleMwangi, Peter N., Robyn-Lee Potgieter, Jeannine Uwimana, Leon Mutesa, Narcisse Muganga, Didier Murenzi, Lisine Tusiyenge, Jason M. Mwenda, Milton T. Mogotsi, Kebareng Rakau, and et al. 2023. "The Evolution of Post-Vaccine G8P[4] Group a Rotavirus Strains in Rwanda; Notable Variance at the Neutralization Epitope Sites" Pathogens 12, no. 5: 658. https://doi.org/10.3390/pathogens12050658