
Genome Characterization and Spaciotemporal Dispersal Analysis of Bagaza Virus Detected in Portugal, 2021

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. The Study
2.2. RNA Extraction and Viral RNA Detection
2.3. Virus Isolation
2.4. Viral Genomic Sequencing
2.5. Phylogenetic and Spatiotemporal Analyses
2.6. ITV and BAGV Genetic Analyses
3. Results
3.1. Virus Isolation
3.2. Viral Genomic Sequencing
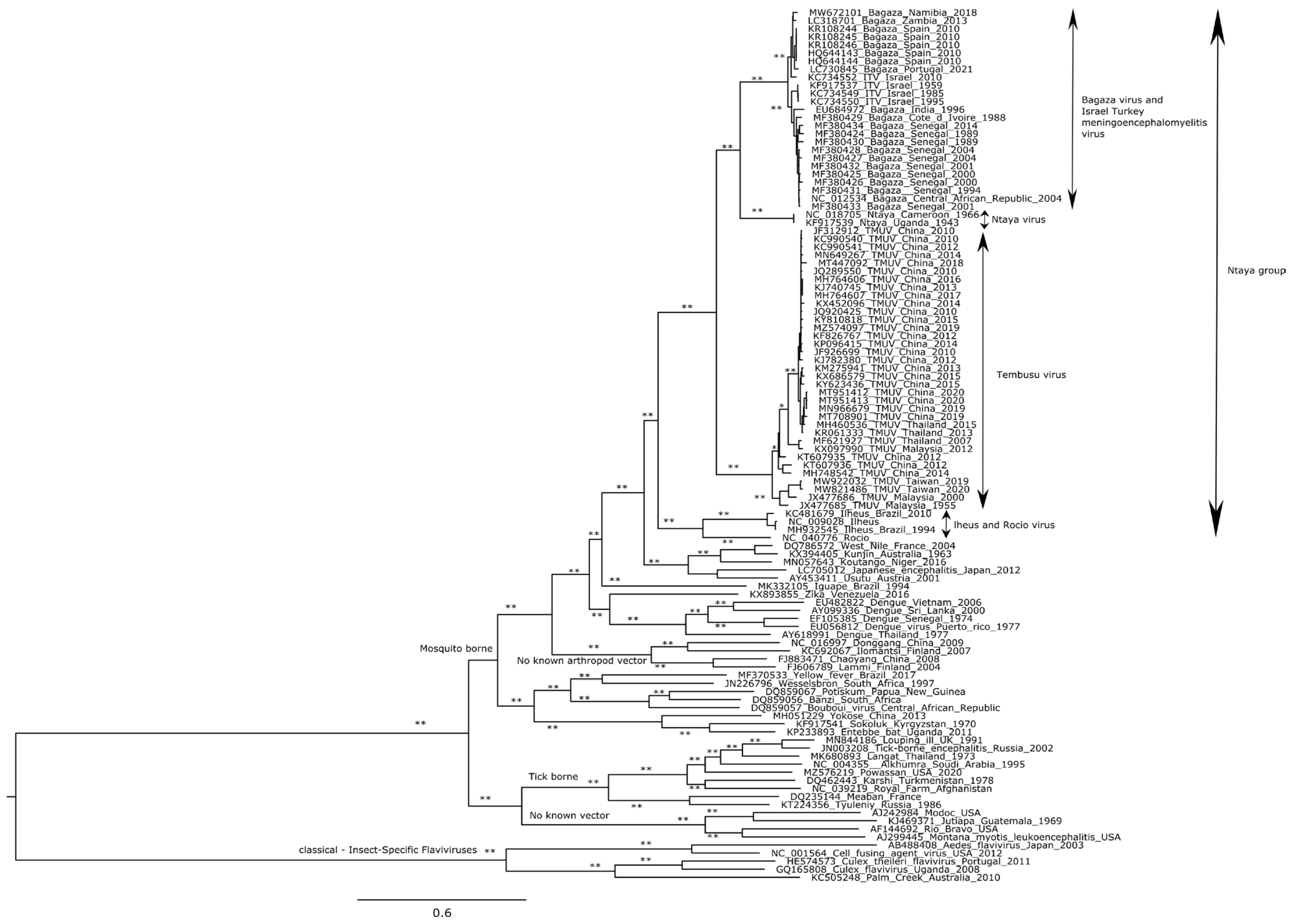
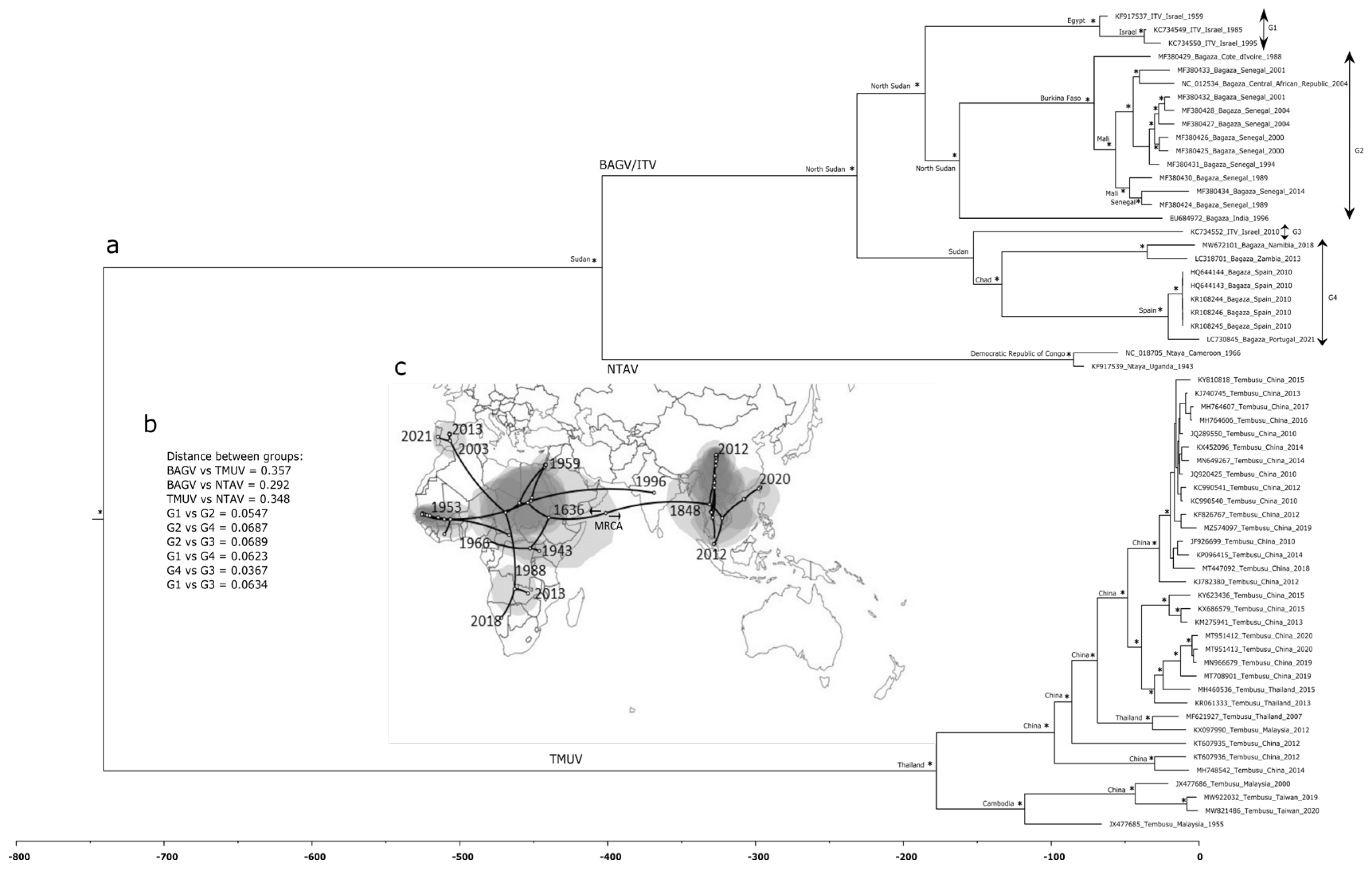
3.3. Phylogenetic and Spatiotemporal Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chong, H.Y.; Leow, C.Y.; Abdul Majeed, A.B.; Leow, C.H. Flavivirus Infection—A Review of Immunopathogenesis, Immunological Response, and Immunodiagnosis. Virus Res. 2019, 274, 197770. [Google Scholar] [CrossRef] [PubMed]
- Behar, A.; Rot, A.; Altory-Natour, A.; Davidson, I. A Two-Branched Upgrade to Demonstrate ITV Transmission by Blood-Sucking Insects. J. Virol. Methods 2021, 296, 114229. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Pinero, J.; Davidson, I.; Elizalde, M.; Perk, S.; Khinich, Y.; Jiménez-Clavero, M.A. Bagaza Virus and Israel Turkey Meningoencephalomyelitis Virus Are a Single Virus Species. J. Gen. Virol. 2014, 95, 883–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Digoutte, J.P. Bagaza (BAG). Am. J. Trop. Med. Hyg. 1978, 27, 376–377. [Google Scholar] [CrossRef]
- Diallo, M.; Nabeth, P.; Ba, K.; Sall, A.A.; Ba, Y.; Mondo, M.; Girault, L.; Abdalahi, M.O.; Mathiot, C. Mosquito Vectors of the 1998-1999 Outbreak of Rift Valley Fever and other Arboviruses (Bagaza, Sanar, Wesselsbron and West Nile) in Mauritania and Senegal. Med. Vet. Entomol. 2005, 19, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Traore-Lamizana, M.; Zeller, H.G.; Mondo, M.; Hervy, J.-P.; Adam, F.; Digoutte, J.-P. Isolations of West Nile and Bagaza Viruses from Mosquitoes (Diptera: Culicidae) in Central Senegal (Ferlo). J. Med. Entomol. 1994, 31, 934–938. [Google Scholar] [CrossRef]
- Bondre, V.P.; Sapkal, G.N.; Yergolkar, P.N.; Fulmali, P.V.; Sankararaman, V.; Ayachit, V.M.; Mishra, A.C.; Gore, M.M. Genetic Characterization of Bagaza Virus (BAGV) Isolated in India and Evidence of Anti-BAGV Antibodies in Sera Collected from Encephalitis Patients. J. Gen. Virol. 2009, 90, 2644–2649. [Google Scholar] [CrossRef]
- Camp, J.V.; Karuvantevida, N.; Chouhna, H.; Safi, E.; Shah, J.N.; Nowotny, N. Mosquito Biodiversity and Mosquito-Borne Viruses in the United Arab Emirates. Parasit. Vectors 2019, 12, 153. [Google Scholar] [CrossRef] [Green Version]
- Agüero, M.; Fernández-Pinero, J.; Buitrago, D.; Sánchez, A.; Elizalde, M.; Miguel, E.S.; Villalba, R.; Llorente, F.; Jiménez-Clavero, M.Á. Bagaza Virus in Partridges and Pheasants, Spain, 2010. Emerg. Infect. Dis. 2011, 17, 1498–1501. [Google Scholar] [CrossRef]
- Steyn, J.; Botha, E.M.; Lourens, C.; Coetzer, J.A.W.; Venter, M. Bagaza Virus in Himalayan Monal Pheasants, South Africa, 2016–2017. Emerg. Infect. Dis. 2019, 25, 2299–2302. [Google Scholar] [CrossRef]
- Höfle, U.; Cardona Cabrera, T.; Sánchez-Cano, A.; Fernández de Mera, I.G.; Risalde, M.A.; Moraga-Fernández, A.; Ortiz, J.A. Bagaza Virus and Plasmodium spp. Coinfection in Red-Legged Partridges (Alectoris Rufa), in Southern Spain 2019. Transbound. Emerg. Dis. 2022, 69, e3393–e3399. [Google Scholar] [CrossRef] [PubMed]
- García-Bocanegra, I.; Zorrilla, I.; Rodríguez, E.; Rayas, E.; Camacho, L.; Redondo, I.; Gómez-Guillamón, F. Monitoring of the Bagaza Virus Epidemic in Wild Bird Species in Spain, 2010. Transbound. Emerg. Dis. 2013, 60, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Dávila, S.G.; Cuenca, O.T.; Gil, M.G.; Toledano-Díaz, O.A.; Castaño, C.; Santiago-Moreno, J.; Campo, J.L. Anti-Predator Behavior in Pure and Hybrid Red-Legged Partridges. Poult. Sci. 2019, 98, 3085–3092. [Google Scholar] [CrossRef] [PubMed]
- Millán, J. Diseases of the Red-Legged Partridge (Alectoris rufa L.): A Review. Wildl. Biol. Pract. 2009, 5, 70–88. [Google Scholar] [CrossRef]
- BirdLife International. European Red List of Birds 2021; Publications Office of the European Union: Luxembourg, 2021. [Google Scholar] [CrossRef]
- Llorente, F.; Pérez-Ramírez, E.; Fernández-Pinero, J.; Elizalde, M.; Figuerola, J.; Soriguer, R.C.; Jiménez-Clavero, M.Á. Bagaza Virus Is Pathogenic and Transmitted by Direct Contact in Experimentally Infected Partridges, but Is Not Infectious in House Sparrows and Adult Mice. Vet. Res. 2015, 46, 93. [Google Scholar] [CrossRef] [Green Version]
- Gamino, V.; Gutiérrez-Guzmán, A.V.; Fernández-De-Mera, I.G.; Ortíz, J.A.; Durán-Martín, M.; de La Fuente, J.; Gortázar, C.; Höfle, U. Natural Bagaza Virus Infection in Game Birds in Southern Spain. Vet. Res. 2012, 43, 65. [Google Scholar] [CrossRef] [Green Version]
- Faye, M.; Faye, O.; Diagne, M.M.; Fall, G.; Weidmann, M.; Sembene, M.; Sall, A.A.; Faye, O. Full-Genome Characterization and Genetic Evolution of West African Isolates of Bagaza Virus. Viruses 2018, 10, 193. [Google Scholar] [CrossRef] [Green Version]
- Queiros, J.; Barros, S.C.; Sanchez-Cano, A.; Henriques, A.M.; Fagulha, T.; dos Santos, F.A.; Duarte, M.D.; Fontoura-Goncalves, C.; Goncalves, D.; Rodrigues, M.; et al. Bagaza Virus in Wild Birds, Portugal, 2021. Emerg. Infect. Dis. 2022, 28, 1504–1506. [Google Scholar] [CrossRef]
- Buitrago, D.; Rocha, A.; Tena-Tomás, C.; Vigo, M.; Agüero, M.; Jiménez-Clavero, M.A. Real-Time Fluorogenic Reverse Transcription Polymerase Chain Reaction Assay for the Specific Detection of Bagaza Virus. J. Vet. Diagn. Investig. 2012, 24, 959–963. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H. SplitsTree: Analyzing and Visualizing Evolutionary Data. Bioinformatics 1998, 14, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, B.; García-Bocanegra, I.; Acevedo, P.; Cáceres, G.; Alves, P.C.; Gortázar, C. Stepping up from Wildlife Disease Surveillance to Integrated Wildlife Monitoring in Europe. Res. Vet. Sci. 2022, 144, 149–156. [Google Scholar] [CrossRef]
- Llorente, F.; Pérez-Ramírez, E.; Fernández-Pinero, J.; Soriguer, R.; Figuerola, J.; Jiménez-Clavero, M.Á. Flaviviruses in Game Birds, Southern Spain, 2011–2012. Emerg. Infect. Dis. 2013, 19, 1023–1025. [Google Scholar] [CrossRef]
- Cao, Z.; Zhang, C.; Liu, Y.; Ye, W.; Han, J.; Ma, G.; Zhang, D.; Xu, F.; Gao, X.; Tang, Y.; et al. Tembusu Virus in Ducks, China. Emerg. Infect. Dis. 2011, 17, 1873–1875. [Google Scholar] [CrossRef]
- Tang, Y.; Gao, X.; Diao, Y.; Feng, Q.; Chen, H.; Liu, X.; Ge, P.; Yu, C. Tembusu Virus in Human, China. Transbound. Emerg. Dis. 2013, 60, 193–196. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Primer | Sequences (5′–3′) | Tm * (°C) | Amplicon Size (bp) |
|---|---|---|---|
| BAGV1F | ACTTTGTGATTGACAGCTCAA | 53 | 626 |
| BAGV626R | GATCATACCCATCCTCTAGCTT | 55 | |
| BAGV516F | TCCCAACTGCTGGAGGAAA | 58 | 682 |
| BAGV1197R | GCTTTGGTGTTGTGGGCCT | 61 | |
| BAGV992F | GGAGTTGAGTGGATTGATGTT | 54 | 668 |
| BAGV1659R | AGTGATTCTCTGTTCTGCC | 53 | |
| BAGV1527F | GGATGGACATGAGCCAGTTTTA | 56 | 752 |
| BAGV2278R | CACTTGGTGTATTCCTTTGC | 53 | |
| BAGV2129F | CAATGGCACAAGAGTGGAAG | 55 | 869 |
| BAGV2997R | ATCACGGCCGTGTCACATTC | 60 | |
| BAGV2771F | GAGGAATTGGAATACGGGTG | 55 | 894 |
| BAGV3664R | CATGTCCCTGTAAGTTATCC | 51 | |
| BAGV3575F | CGGAAAAGATGGACAGGCCGGG | 65 | 696 |
| BAGV4271R | CCATGTCTCCTTCGTCAAAGTGT | 58 | |
| BAGV4081F | GAAAGGTGGAGTGCTGATTG | 55 | 740 |
| BAGV4820R | GTCCTCCATACGATATCAAGTC | 53 | |
| BAGV4688F | GTAGGAGTGATGTTTGATGG | 51 | 745 |
| BAGV5432R | CATCCATCACAAACAAGTTGT | 52 | |
| BAGV5262F | CAGCTGAGATAGCGGAAGCT | 59 | 850 |
| BAGV6111R | GGCTCATAGAGTTGGGCTAC | 57 | |
| BAGV5944F | GCAGAGACGCGGAAGAATTG | 58 | 881 |
| BAGV6824R | TGTCAGTTTGTGATCTCTGTC | 53 | |
| BAGV6725F | GAAGTCCAACCACAAAAGATAG | 52 | 924 |
| BAGV7649R | GCCTCCTCTTCTCATGCTTC | 57 | |
| BAGV7552F | CCATGTGCCATCTAATGAGGAAG | 57 | 999 |
| BAGV8550R | TGTATGGATTTTCTGTGTCAT | 50 | |
| BAGV8313F | GGAGCCAGTGGGAACATCAC | 60 | 773 |
| BAGV9085R | AGCCACATATACCATATGGCCC | 58 | |
| BAGV8988F | TGTGAGACATGCATCTACAA | 52 | 721 |
| BAGV9708R | TGCTGTTCAAGAAATGCAATG | 53 | |
| BAGV9598F | TGGGCAAGAATGGAAGAGA | 55 | 839 |
| BAGV10436R | CTACTTACTTACTTAAATCTGATTA | 45 | |
| BAGV10237F | CAGAGAACATATACACACCAAT | 50 | 560 |
| BAGV10796R | GGGGTCTCCTCTAACCTCTA | 56 |
| Proteins | Size a | BAGV/Spain HQ644143 | ITV KC734550 | NTAV NC018705 | TMUV MN649267 |
|---|---|---|---|---|---|
| ID% | ID% | ID% | ID% | ||
| C | 122 | 96.68 | 95.08 | 80.33 | 70.43 |
| preM | 167 | 99.40 | 100.00 | 87.43 | 82.42 |
| E | 501 | 99.60 | 98.00 | 92.02 | 83.43 |
| NS1 | 352 | 99.15 | 98.01 | 83.52 | 77.25 |
| NS2a | 227 | 99.12 | 97.80 | 76.21 | 63.88 |
| NS2b | 131 | 100.00 | 100.00 | 85.50 | 75.57 |
| NS3 | 619 | 99.35 | 98.87 | 91.44 | 85.92 |
| NS4a | 127 | 100.00 | 100.00 | 91.34 | 82.68 |
| 2K | 22 | 100.00 | 95.45 | 86.36 | 90.91 |
| NS4b | 254 | 98.82 | 98.82 | 87.01 | 83.46 |
| NS5 | 905 | 99.67 | 99.01 | 90.83 | 85.86 |
| Amino Acid Position | Protein | 1 ITV/2 BAGVSpain/3 BAGVP |
|---|---|---|
| 83 | C | K/K/R |
| 100 | C | * G/G/S 1 |
| 659 | E | K/K/R |
| 699 | E | * S/S/F 2 |
| 838 | NS1 | E/E/D |
| 896 | NS1 | * W/W/L 3 |
| 1051 | NS1 | K/K/R |
| 1202 | NS2a | * L/L/M 4 |
| 1547 | NS3 | H/H/R |
| 1885 | NS3 | * Q/Q/P 5 |
| 1887 | NS3 | * N/N/Y 6 |
| 2283 | NS4b | * S/S/N 7 |
| 2358 | NS4b | * I/I/T 1 |
| 2435 | NS4b | I/I/V |
| 2703 | NS5 | I/I/V |
| 3286 | NS5 | * G/G/S 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falcão, M.; Barros, M.; Duarte, M.D.; Santos, F.A.d.; Fagulha, T.; Henriques, M.; Ramos, F.; Duarte, A.; Luís, T.; Parreira, R.; et al. Genome Characterization and Spaciotemporal Dispersal Analysis of Bagaza Virus Detected in Portugal, 2021. Pathogens 2023, 12, 150. https://doi.org/10.3390/pathogens12020150
Falcão M, Barros M, Duarte MD, Santos FAd, Fagulha T, Henriques M, Ramos F, Duarte A, Luís T, Parreira R, et al. Genome Characterization and Spaciotemporal Dispersal Analysis of Bagaza Virus Detected in Portugal, 2021. Pathogens. 2023; 12(2):150. https://doi.org/10.3390/pathogens12020150
Chicago/Turabian StyleFalcão, Marta, Margarida Barros, Margarida D. Duarte, Fábio Abade dos Santos, Teresa Fagulha, Margarida Henriques, Fernanda Ramos, Ana Duarte, Tiago Luís, Ricardo Parreira, and et al. 2023. "Genome Characterization and Spaciotemporal Dispersal Analysis of Bagaza Virus Detected in Portugal, 2021" Pathogens 12, no. 2: 150. https://doi.org/10.3390/pathogens12020150