
How to Inhibit Nuclear Factor-Kappa B Signaling: Lessons from Poxviruses
Abstract
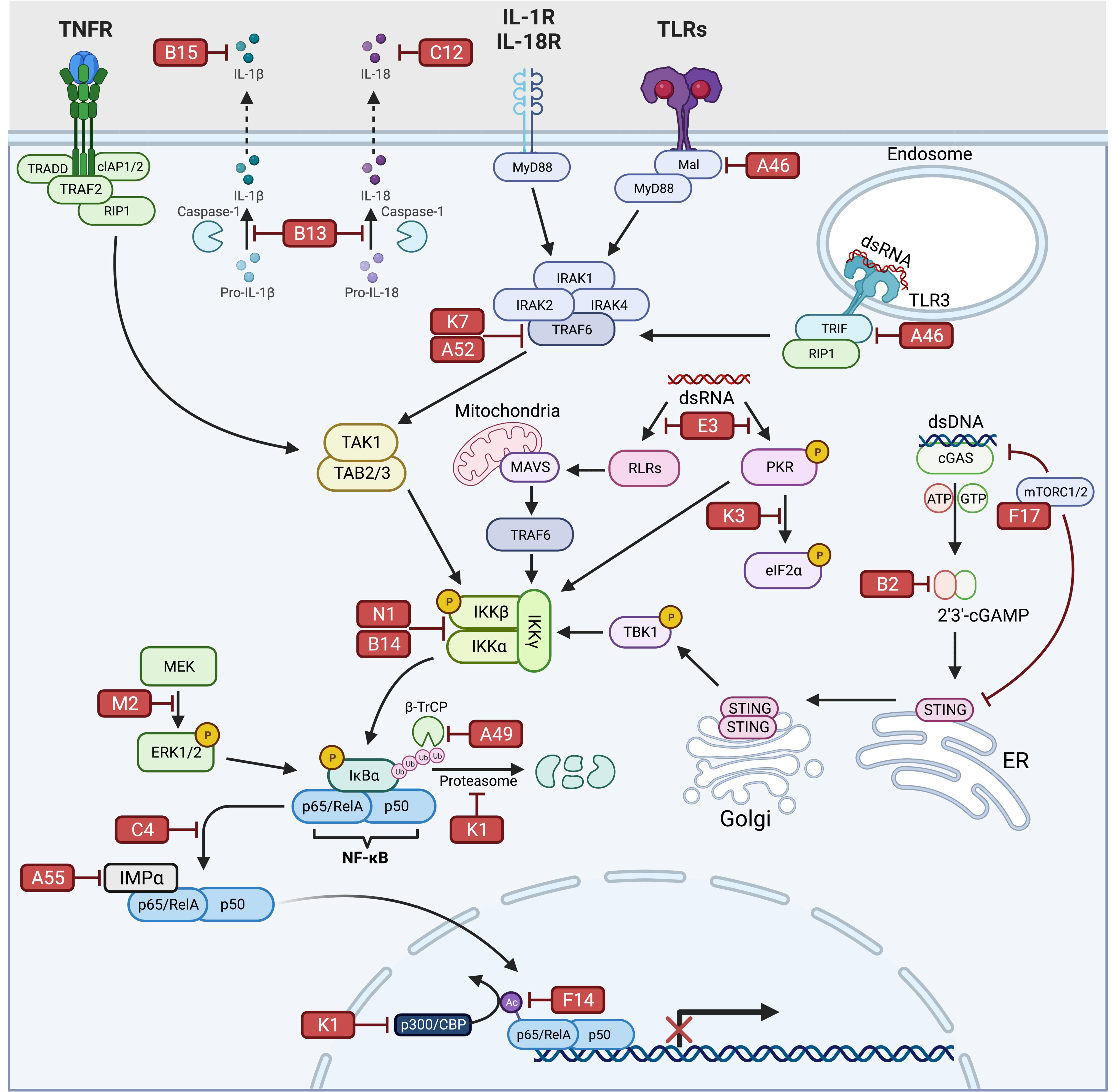
:1. Introduction
2. NF-κB Signaling
3. VV Inhibitors Targeting Receptors Mediating NF-κB Activation
3.1. E3
3.2. K3
3.3. K1
3.4. C12
3.5. B15
4. VV Inhibitors Targeting NF-κB Signaling Intermediates
4.1. K7
4.2. A46
4.3. A52
4.4. B14
4.5. N1
4.6. B13
4.7. B2
4.8. F17
5. VV Inhibitors Directly Targeting NF-κB Complex Activation/Activity
5.1. A49
5.2. F14
5.3. K1
5.4. A55
5.5. C4
5.6. M2
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maclachlan, N.J.; Dubovi, E.J.; Barthold, S.W.; Swayne, D.E.; Winton, J.R. Chapter 7—Poxviridae, 15th ed.; Elsevier: Amsterdam, The Netherlands; Academic Press: Cambridge, MA, USA, 2017; 581p. [Google Scholar] [CrossRef]
- Becker, M.N.; Moyer, R.W. Entomopoxviruses, 3rd ed.; Mahy, B.W.J., Van Regenmortel, M.H.V., Eds.; Academic Press: Cambridge, MA, USA, 2008. [Google Scholar] [CrossRef]
- McFadden, G. Poxvirus tropism. Nat. Rev. Microbiol. 2005, 3, 201–213. [Google Scholar] [CrossRef]
- Theves, C.; Crubezy, E.; Biagini, P. History of Smallpox and Its Spread in Human Populations. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. Monkeypox: A potential global threat? J. Med. Virol. 2022, 94, 4034–4036. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, R.K.; Tuli, H.S.; Sarangi, A.K.; Chakraborty, S.; Chandran, D.; Chakraborty, C.; Dhama, K. Unexpected sudden rise of human monkeypox cases in multiple non-endemic countries amid COVID-19 pandemic and salient counteracting strategies: Another potential global threat? Int. J. Surg. 2022, 103, 106705. [Google Scholar] [CrossRef]
- Hamdi, J.; Munyanduki, H.; Omari Tadlaoui, K.; El Harrak, M.; Fassi Fihri, O. Capripoxvirus Infections in Ruminants: A Review. Microorganisms 2021, 9, 902. [Google Scholar] [CrossRef] [PubMed]
- Conrad, S.J.; Liu, J. Poxviruses as Gene Therapy Vectors: Generating Poxviral Vectors Expressing Therapeutic Transgenes. Methods Mol. Biol. 2019, 1937, 189–209. [Google Scholar] [CrossRef] [PubMed]
- Ricordel, M.; Foloppe, J.; Pichon, C.; Findeli, A.; Tosch, C.; Cordier, P.; Cochin, S.; Quemeneur, E.; Camus-Bouclainville, C.; Bertagnoli, S.; et al. Oncolytic properties of non-vaccinia poxviruses. Oncotarget 2018, 9, 35891–35906. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Bruneau, R.C.; Brennan, G.; Rothenburg, S. Battle Royale: Innate Recognition of Poxviruses and Viral Immune Evasion. Biomedicines 2021, 9, 765. [Google Scholar] [CrossRef] [PubMed]
- Struzik, J.; Szulc-Dabrowska, L. NF-kappaB as an Important Factor in Optimizing Poxvirus-Based Vaccines against Viral Infections. Pathogens 2020, 9, 1001. [Google Scholar] [CrossRef] [PubMed]
- Chiuppesi, F.; Salazar, M.D.; Contreras, H.; Nguyen, V.; Martinez, J.; Park, S.; Nguyen, J.; Kha, M.; Iniguez, A.; Zhou, Q.; et al. Development of a Multi-Antigenic SARS-CoV-2 Vaccine Using a Synthetic Poxvirus Platform. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Lazaro-Frias, A.; Gomez-Medina, S.; Sanchez-Sampedro, L.; Ljungberg, K.; Ustav, M.; Liljestrom, P.; Munoz-Fontela, C.; Esteban, M.; Garcia-Arriaza, J. Distinct Immunogenicity and Efficacy of Poxvirus-Based Vaccine Candidates against Ebola Virus Expressing GP and VP40 Proteins. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-F.; Greene, W.C. Shaping the nuclear action of NF-κB. Nat. Rev. Mol. Cell Biol. 2004, 5, 392–401. [Google Scholar] [CrossRef]
- Siebenlist, U.; Franzoso, G.; Brown, K. Structure, Regulation and Function of NF-kappaB. Annu. Rev. Cell Biol. 1994, 10, 405–455. [Google Scholar] [CrossRef] [PubMed]
- Palombella, V.J.; Rando, O.J.; Goldberg, A.L.; Maniatis, T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 1994, 78, 773–785. [Google Scholar] [CrossRef]
- Savinova, O.V.; Hoffmann, A.; Ghosh, G. The Nfkb1 and Nfkb2 Proteins p105 and p100 Function as the Core of High-Molecular-Weight Heterogeneous Complexes. Mol. Cell 2009, 34, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Rice, N.R.; MacKichan, M.L.; Israel, A. The precursor of NF-kappa B p50 has I kappa B-like functions. Cell 1992, 71, 243–253. [Google Scholar] [CrossRef]
- Sun, S.-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef]
- Jin, J.; Hu, H.; Li, H.S.; Yu, J.; Xiao, Y.; Brittain, G.C.; Zou, Q.; Cheng, X.; Mallette, F.A.; Watowich, S.S.; et al. Noncanonical NF-kappaB pathway controls the production of type I interferons in antiviral innate immunity. Immunity 2014, 40, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, K.; Garofalo, R.P.; Brasier, A.R. Respiratory syncytial virus induces RelA release from cytoplasmic 100-kDa NF-kappa B2 complexes via a novel retinoic acid-inducible gene-I{middle dot}NF- kappa B-inducing kinase signaling pathway. J. Biol. Chem. 2008, 283, 23169–23178. [Google Scholar] [CrossRef] [PubMed]
- Manches, O.; Fernandez, M.V.; Plumas, J.; Chaperot, L.; Bhardwaj, N. Activation of the noncanonical NF-kappaB pathway by HIV controls a dendritic cell immunoregulatory phenotype. Proc. Natl. Acad. Sci. USA 2012, 109, 14122–14127. [Google Scholar] [CrossRef] [PubMed]
- Ruckle, A.; Haasbach, E.; Julkunen, I.; Planz, O.; Ehrhardt, C.; Ludwig, S. The NS1 protein of influenza A virus blocks RIG-I-mediated activation of the noncanonical NF-kappaB pathway and p52/RelB-dependent gene expression in lung epithelial cells. J. Virol. 2012, 86, 10211–10217. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Chang, Y.; Wei, W. The role of BAFF in the progression of rheumatoid arthritis. Cytokine 2015, 76, 537–544. [Google Scholar] [CrossRef]
- Brightbill, H.D.; Jackman, J.K.; Suto, E.; Kennedy, H.; Jones, C., 3rd; Chalasani, S.; Lin, Z.; Tam, L.; Roose-Girma, M.; Balazs, M.; et al. Conditional Deletion of NF-kappaB-Inducing Kinase (NIK) in Adult Mice Disrupts Mature B Cell Survival and Activation. J. Immunol. 2015, 195, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.; Macht, A.; Waisman, A.; Hovelmeyer, N. NF-kappaB-inducing kinase is essential for B-cell maintenance in mice. Eur. J. Immunol. 2016, 46, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Valino-Rivas, L.; Gonzalez-Lafuente, L.; Sanz, A.B.; Ruiz-Ortega, M.; Ortiz, A.; Sanchez-Nino, M.D. Non-canonical NFkappaB activation promotes chemokine expression in podocytes. Sci. Rep. 2016, 6, 28857. [Google Scholar] [CrossRef]
- Carragher, D.; Johal, R.; Button, A.; White, A.; Eliopoulos, A.; Jenkinson, E.; Anderson, G.; Caamano, J. A stroma-derived defect in NF-kappaB2-/- mice causes impaired lymph node development and lymphocyte recruitment. J. Immunol. 2004, 173, 2271–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, J.C.; Basak, S.; James, E.S.; Quiambo, R.S.; Kinsella, M.C.; Alegre, M.L.; Weih, F.; Franzoso, G.; Hoffmann, A.; Fu, Y.X. Coordination between NF-kappaB family members p50 and p52 is essential for mediating LTbetaR signals in the development and organization of secondary lymphoid tissues. Blood 2006, 107, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, X.; Nakaya, M.; Jin, W.; Cheng, X.; Sun, S.C. T cell-intrinsic function of the noncanonical NF-kappaB pathway in the regulation of GM-CSF expression and experimental autoimmune encephalomyelitis pathogenesis. J. Immunol. 2014, 193, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Zhou, X.; Xie, X.; Chen, X.; Jie, Z.; Zou, Q.; Hu, H.; Zhu, L.; Cheng, X.; et al. Cell intrinsic role of NF-kappaB-inducing kinase in regulating T cell-mediated immune and autoimmune responses. Sci. Rep. 2016, 6, 22115. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C.; Ley, S.C. New insights into NF-κB regulation and function. Trends Immunol. 2008, 29, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Ting, A.T.; Bertrand, M.J.M. More to Life than NF-kappaB in TNFR1 Signaling. Trends Immunol. 2016, 37, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Ruben, S.M.; Scheinman, R.I.; Haskill, S.; Rosen, C.A.; Baldwin, A.S. I kappa B interacts with the nuclear localization sequences of the subunits of NF-kappa B: A mechanism for cytoplasmic retention. Genes Dev. 1992, 6, 1899–1913. [Google Scholar] [CrossRef]
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The SCFbeta -TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in Ikappa Balpha and beta -catenin and stimulates Ikappa Balpha ubiquitination in vitro. Genes Dev. 1999, 13, 270–283. [Google Scholar] [CrossRef]
- Keating, S.E.; Maloney, G.M.; Moran, E.M.; Bowie, A.G. IRAK-2 Participates in Multiple Toll-like Receptor Signaling Pathways to NFκB via Activation of TRAF6 Ubiquitination. J. Biol. Chem. 2007, 282, 33435–33443. [Google Scholar] [CrossRef]
- Landstrom, M. The TAK1-TRAF6 signalling pathway. Int. J. Biochem. Cell Biol. 2010, 42, 585–589. [Google Scholar] [CrossRef]
- Zamanian-Daryoush, M.; Mogensen, T.H.; DiDonato, J.A.; Williams, B.R. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol. Cell Biol. 2000, 20, 1278–1290. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.X.; Liu, Z.X.; Sun, Y.P.; Zhu, J.; Lu, S.Y.; Liu, X.S.; Huang, Q.H.; Xie, Y.Y.; Zhu, H.B.; Dang, S.Y.; et al. Rig-I regulates NF-kappaB activity through binding to Nf-kappab1 3'-UTR mRNA. Proc. Natl. Acad. Sci. USA 2013, 110, 6459–6464. [Google Scholar] [CrossRef]
- Yoneyama, M.; Fujita, T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 2009, 227, 54–65. [Google Scholar] [CrossRef]
- Roff, M.; Thompson, J.; Rodriguez, M.S.; Jacque, J.M.; Baleux, F.; Arenzana-Seisdedos, F.; Hay, R.T. Role of IkappaBalpha ubiquitination in signal-induced activation of NFkappaB in vivo. J. Biol. Chem. 1996, 271, 7844–7850. [Google Scholar] [CrossRef]
- Deng, J.; Lu, P.D.; Zhang, Y.; Scheuner, D.; Kaufman, R.J.; Sonenberg, N.; Harding, H.P.; Ron, D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell Biol. 2004, 24, 10161–10168. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Y.; Wek, S.A.; McGrath, B.C.; Scheuner, D.; Kaufman, R.J.; Cavener, D.R.; Wek, R.C. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol. Cell Biol. 2003, 23, 5651–5663. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein that Activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef]
- Fang, R.; Wang, C.; Jiang, Q.; Lv, M.; Gao, P.; Yu, X.; Mu, P.; Zhang, R.; Bi, S.; Feng, J.M.; et al. NEMO-IKKbeta Are Essential for IRF3 and NF-kappaB Activation in the cGAS-STING Pathway. J. Immunol. 2017, 199, 3222–3233. [Google Scholar] [CrossRef]
- Chen, L.F. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002, 21, 6539–6548. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef]
- Zhang, G.; Ghosh, S. Toll-like receptor-mediated NF-kappaB activation: A phylogenetically conserved paradigm in innate immunity. J. Clin. Investig. 2001, 107, 13–19. [Google Scholar] [CrossRef]
- Moriuchi, H.; Moriuchi, M.; Fauci, A.S. Nuclear factor-kappa B potently up-regulates the promoter activity of RANTES, a chemokine that blocks HIV infection. J. Immunol. 1997, 158, 3483–3491. [Google Scholar] [PubMed]
- Catz, S.D.; Johnson, J.L. Transcriptional regulation of bcl-2 by nuclear factor κB and its significance in prostate cancer. Oncogene 2001, 20, 7342–7351. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.A.; Heilbock, C.; Kassel, O.; Frossard, N. Transcription of stem cell factor (SCF) is potentiated by glucocorticoids and interleukin-1β through concerted regulation of a GRE-like and an NF-κB response element. FASEB J. 2003, 17, 2334–2336. [Google Scholar] [CrossRef]
- Sadler, A.J.; Williams, B.R.G. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Lawler, C.; Brady, G. Poxviral Targeting of Interferon Regulatory Factor Activation. Viruses 2020, 12, 1191. [Google Scholar] [CrossRef] [PubMed]
- Szczerba, M.; Subramanian, S.; Trainor, K.; McCaughan, M.; Kibler, K.V.; Jacobs, B.L. Small Hero with Great Powers: Vaccinia Virus E3 Protein and Evasion of the Type I IFN Response. Biomedicines 2022, 10, 235. [Google Scholar] [CrossRef]
- Smith, G.L.; Talbot-Cooper, C.; Lu, Y. How Does Vaccinia Virus Interfere With Interferon? Adv. Virus Res. 2018, 100, 355–378. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Beg, A.A. Defining Emerging Roles for NF-κB in Antivirus Responses: Revisiting the Interferon-β Enhanceosome Paradigm. PLoS Pathog. 2011, 7, e1002165. [Google Scholar] [CrossRef] [Green Version]
- Basagoudanavar, S.H.; Thapa, R.J.; Nogusa, S.; Wang, J.; Beg, A.A.; Balachandran, S. Distinct roles for the NF-kappa B RelA subunit during antiviral innate immune responses. J. Virol. 2011, 85, 2599–2610. [Google Scholar] [CrossRef]
- Pfeffer, L.M. The role of nuclear factor kappaB in the interferon response. J. Interferon Cytokine Res. 2011, 31, 553–559. [Google Scholar] [CrossRef]
- Silva, T.; Temerozo, J.R.; do Vale, G.; Ferreira, A.C.; Soares, V.C.; Dias, S.S.G.; Sardella, G.; Bou-Habib, D.C.; Siqueira, M.; Souza, T.M.L.; et al. The Chemokine CCL5 Inhibits the Replication of Influenza A Virus Through SAMHD1 Modulation. Front. Cell Infect. Microbiol. 2021, 11, 549020. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, L.M.; Kim, J.-G.; Pfeffer, S.R.; Carrigan, D.J.; Baker, D.P.; Wei, L.; Homayouni, R. Role of Nuclear Factor-κB in the Antiviral Action of Interferon and Interferon-regulated Gene Expression. J. Biol. Chem. 2004, 279, 31304–31311. [Google Scholar] [CrossRef] [PubMed]
- Morales, D.J.; Lenschow, D.J. The antiviral activities of ISG15. J. Mol. Biol. 2013, 425, 4995–5008. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Katoh, H.; Kayama, H.; Saiga, H.; Okuyama, M.; Okamoto, T.; Umemoto, E.; Matsuura, Y.; Yamamoto, M.; Takeda, K. Ifit1 inhibits Japanese encephalitis virus replication through binding to 5' capped 2'-O unmethylated RNA. J. Virol. 2013, 87, 9997–10003. [Google Scholar] [CrossRef]
- Lu, R.; Moore, P.A.; Pitha, P.M. Stimulation of IRF-7 gene expression by tumor necrosis factor alpha: Requirement for NFkappa B transcription factor and gene accessibility. J. Biol. Chem. 2002, 277, 16592–16598. [Google Scholar] [CrossRef]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2018, 11, 480. [Google Scholar] [CrossRef]
- Cesaro, T.; Michiels, T. Inhibition of PKR by Viruses. Front. Microbiol. 2021, 12, 757238. [Google Scholar] [CrossRef]
- Willis, K.L.; Langland, J.O.; Shisler, J.L. Viral double-stranded RNAs from vaccinia virus early or intermediate gene transcripts possess PKR activating function, resulting in NF-kappaB activation, when the K1 protein is absent or mutated. J. Biol. Chem. 2011, 286, 7765–7778. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.W.; Uribe, L.H.; Jacobs, B.L. Rescue of vaccinia virus lacking the E3L gene by mutants of E3L. J. Virol. 1995, 69, 6605–6608. [Google Scholar] [CrossRef]
- Beattie, E.; Paoletti, E.; Tartaglia, J. Distinct patterns of IFN sensitivity observed in cells infected with vaccinia K3L- and E3L- mutant viruses. Virology 1995, 210, 254–263. [Google Scholar] [CrossRef]
- Romano, P.R.; Zhang, F.; Tan, S.L.; Garcia-Barrio, M.T.; Katze, M.G.; Dever, T.E.; Hinnebusch, A.G. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: Role of complex formation and the E3 N-terminal domain. Mol. Cell Biol. 1998, 18, 7304–7316. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.W.; Watson, J.C.; Jacobs, B.L. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1992, 89, 4825–4829. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.C.; Chang, H.W.; Jacobs, B.L. Characterization of a vaccinia virus-encoded double-stranded RNA-binding protein that may be involved in inhibition of the double-stranded RNA-dependent protein kinase. Virology 1991, 185, 206–216. [Google Scholar] [CrossRef]
- Sharp, T.V.; Moonan, F.; Romashko, A.; Joshi, B.; Barber, G.N.; Jagus, R. The vaccinia virus E3L gene product interacts with both the regulatory and the substrate binding regions of PKR: Implications for PKR autoregulation. Virology 1998, 250, 302–315. [Google Scholar] [CrossRef]
- Liu, R.; Moss, B. Opposing Roles of Double-Stranded RNA Effector Pathways and Viral Defense Proteins Revealed with CRISPR-Cas9 Knockout Cell Lines and Vaccinia Virus Mutants. J. Virol. 2016, 90, 7864–7879. [Google Scholar] [CrossRef]
- Deng, L.; Dai, P.; Parikh, T.; Cao, H.; Bhoj, V.; Sun, Q.; Chen, Z.; Merghoub, T.; Houghton, A.; Shuman, S. Vaccinia virus subverts a mitochondrial antiviral signaling protein-dependent innate immune response in keratinocytes through its double-stranded RNA binding protein, E3. J. Virol. 2008, 82, 10735–10746. [Google Scholar] [CrossRef] [PubMed]
- Brandt, T.A.; Jacobs, B.L. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J. Virol. 2001, 75, 850–856. [Google Scholar] [CrossRef]
- Shors, T.; Kibler, K.V.; Perkins, K.B.; Seidler-Wulff, R.; Banaszak, M.P.; Jacobs, B.L. Complementation of vaccinia virus deleted of the E3L gene by mutants of E3L. Virology 1997, 239, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Bahar, M.W.; Graham, S.C.; Chen, R.A.; Cooray, S.; Smith, G.L.; Stuart, D.I.; Grimes, J.M. How vaccinia virus has evolved to subvert the host immune response. J. Struct. Biol. 2011, 175, 127–134. [Google Scholar] [CrossRef]
- Carroll, K.; Elroy-Stein, O.; Moss, B.; Jagus, R. Recombinant vaccinia virus K3L gene product prevents activation of double-stranded RNA-dependent, initiation factor 2 alpha-specific protein kinase. J. Biol. Chem. 1993, 268, 12837–12842. [Google Scholar] [CrossRef]
- Davies, M.V.; Elroy-Stein, O.; Jagus, R.; Moss, B.; Kaufman, R.J. The vaccinia virus K3L gene product potentiates translation by inhibiting double-stranded-RNA-activated protein kinase and phosphorylation of the alpha subunit of eukaryotic initiation factor 2. J. Virol. 1992, 66, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.C.; Sicheri, F. X-ray crystal structure and functional analysis of vaccinia virus K3L reveals molecular determinants for PKR subversion and substrate recognition. Mol. Cell 2002, 10, 295–305. [Google Scholar] [CrossRef]
- Langland, J.O.; Jacobs, B.L. The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology 2002, 299, 133–141. [Google Scholar] [CrossRef]
- Elde, N.C.; Child, S.J.; Geballe, A.P.; Malik, H.S. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 2009, 457, 485–489. [Google Scholar] [CrossRef]
- Park, C.; Peng, C.; Rahman, M.J.; Haller, S.L.; Tazi, L.; Brennan, G.; Rothenburg, S. Orthopoxvirus K3 orthologs show virus- and host-specific inhibition of the antiviral protein kinase PKR. PLoS Pathog. 2021, 17, e1009183. [Google Scholar] [CrossRef]
- Rice, A.D.; Turner, P.C.; Embury, J.E.; Moldawer, L.L.; Baker, H.V.; Moyer, R.W. Roles of vaccinia virus genes E3L and K3L and host genes PKR and RNase L during intratracheal infection of C57BL/6 mice. J. Virol. 2011, 85, 550–567. [Google Scholar] [CrossRef] [PubMed]
- Shisler, J.L.; Jin, X.L. The vaccinia virus K1L gene product inhibits host NF-kappaB activation by preventing IkappaBalpha degradation. J. Virol. 2004, 78, 3553–3560. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Mahajan, A.; Tsai, M.D. Ankyrin repeat: A unique motif mediating protein-protein interactions. Biochemistry 2006, 45, 15168–15178. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Meng, X.; Xiang, Y.; Deng, J. Structure function studies of vaccinia virus host range protein k1 reveal a novel functional surface for ankyrin repeat proteins. J. Virol. 2010, 84, 3331–3338. [Google Scholar] [CrossRef]
- Bravo Cruz, A.G.; Han, A.; Roy, E.J.; Guzman, A.B.; Miller, R.J.; Driskell, E.A.; O’Brien, W.D., Jr.; Shisler, J.L. Deletion of the K1L Gene Results in a Vaccinia Virus That Is Less Pathogenic Due to Muted Innate Immune Responses, yet Still Elicits Protective Immunity. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Symons, J.A.; Adams, E.; Tscharke, D.C.; Reading, P.C.; Waldmann, H.; Smith, G.L. The vaccinia virus C12L protein inhibits mouse IL-18 and promotes virus virulence in the murine intranasal model. J. Gen. Virol. 2002, 83, 2833–2844. [Google Scholar] [CrossRef] [PubMed]
- Stylianou, E. Interleukins|IL-1 and IL-18. Encycl. Respir. Med. 2006, 23, 350–354. [Google Scholar] [CrossRef]
- Smith, V.P.; Bryant, N.A.; Alcami, A. Ectromelia, vaccinia and cowpox viruses encode secreted interleukin-18-binding proteins. J. Gen. Virol. 2000, 81, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Krumm, B.; Meng, X.; Li, Y.; Xiang, Y.; Deng, J. Structural basis for antagonism of human interleukin 18 by poxvirus interleukin 18-binding protein. Proc. Natl. Acad. Sci. USA 2008, 105, 20711–20715. [Google Scholar] [CrossRef] [PubMed]
- Reading, P.C.; Smith, G.L. Vaccinia virus interleukin-18-binding protein promotes virulence by reducing gamma interferon production and natural killer and T-cell activity. J. Virol. 2003, 77, 9960–9968. [Google Scholar] [CrossRef]
- Jacobs, B.L.; Langland, J.O.; Kibler, K.V.; Denzler, K.L.; White, S.D.; Holechek, S.A.; Wong, S.; Huynh, T.; Baskin, C.R. Vaccinia virus vaccines: Past, present and future. Antivir. Res. 2009, 84, 1–13. [Google Scholar] [CrossRef]
- Moutaftsi, M.; Bui, H.H.; Peters, B.; Sidney, J.; Salek-Ardakani, S.; Oseroff, C.; Pasquetto, V.; Crotty, S.; Croft, M.; Lefkowitz, E.J.; et al. Vaccinia virus-specific CD4+ T cell responses target a set of antigens largely distinct from those targeted by CD8+ T cell responses. J. Immunol. 2007, 178, 6814–6820. [Google Scholar] [CrossRef]
- Smith, G.L.; Chan, Y.S. Two vaccinia virus proteins structurally related to the interleukin-1 receptor and the immunoglobulin superfamily. J. Gen. Virol. 1991, 72 Pt 3, 511–518. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Spriggs, M.K.; Hruby, D.E.; Maliszewski, C.R.; Pickup, D.J.; Sims, J.E.; Buller, R.M.; VanSlyke, J. Vaccinia and cowpox viruses encode a novel secreted interleukin-1-binding protein. Cell 1992, 71, 145–152. [Google Scholar] [CrossRef]
- Alcami, A.; Smith, G.L. A soluble receptor for interleukin-1 beta encoded by vaccinia virus: A novel mechanism of virus modulation of the host response to infection. Cell 1992, 71, 153–167. [Google Scholar] [CrossRef]
- Benfield, C.T.O.; Ren, H.; Lucas, S.J.; Bahsoun, B.; Smith, G.L. Vaccinia virus protein K7 is a virulence factor that alters the acute immune response to infection. J. Gen. Virol. 2013, 94, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Kalverda, A.P.; Thompson, G.S.; Vogel, A.; Schroder, M.; Bowie, A.G.; Khan, A.R.; Homans, S.W. Poxvirus K7 protein adopts a Bcl-2 fold: Biochemical mapping of its interactions with human DEAD box RNA helicase DDX3. J. Mol. Biol. 2009, 385, 843–853. [Google Scholar] [CrossRef]
- Schroder, M.; Baran, M.; Bowie, A.G. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J. 2008, 27, 2147–2157. [Google Scholar] [CrossRef]
- Stack, J.; Haga, I.R.; Schroder, M.; Bartlett, N.W.; Maloney, G.; Reading, P.C.; Fitzgerald, K.A.; Smith, G.L.; Bowie, A.G. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J. Exp. Med. 2005, 201, 1007–1018. [Google Scholar] [CrossRef]
- Smith, G.L.; Benfield, C.T.O.; Maluquer de Motes, C.; Mazzon, M.; Ember, S.W.J.; Ferguson, B.J.; Sumner, R.P. Vaccinia virus immune evasion: Mechanisms, virulence and immunogenicity. J. Gen. Virol. 2013, 94, 2367–2392. [Google Scholar] [CrossRef]
- Bowie, A.; Kiss-Toth, E.; Symons, J.A.; Smith, G.L.; Dower, S.K.; O’Neill, L.A. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 2000, 97, 10162–10167. [Google Scholar] [CrossRef] [PubMed]
- Sheedy, F.J.; O’Neill, L.A. The Troll in Toll: Mal and Tram as bridges for TLR2 and TLR4 signaling. J. Leukoc. Biol. 2007, 82, 196–203. [Google Scholar] [CrossRef]
- Burns, K.; Martinon, F.; Esslinger, C.; Pahl, H.; Schneider, P.; Bodmer, J.L.; Di Marco, F.; French, L.; Tschopp, J. MyD88, an adapter protein involved in interleukin-1 signaling. J. Biol. Chem. 1998, 273, 12203–12209. [Google Scholar] [CrossRef]
- Jefferies, C.; Bowie, A.; Brady, G.; Cooke, E.L.; Li, X.; O’Neill, L.A. Transactivation by the p65 subunit of NF-kappaB in response to interleukin-1 (IL-1) involves MyD88, IL-1 receptor-associated kinase 1, TRAF-6, and Rac1. Mol. Cell Biol. 2001, 21, 4544–4552. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, H.; Heo, L.; Seok, C.; Choe, J. Structure of vaccinia virus A46, an inhibitor of TLR4 signaling pathway, shows the conformation of VIPER motif. Protein Sci. 2014, 23, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Fedosyuk, S.; Bezerra, G.A.; Radakovics, K.; Smith, T.K.; Sammito, M.; Bobik, N.; Round, A.; Ten Eyck, L.F.; Djinovic-Carugo, K.; Uson, I.; et al. Vaccinia Virus Immunomodulator A46: A Lipid and Protein-Binding Scaffold for Sequestering Host TIR-Domain Proteins. PLoS Pathog. 2016, 12, e1006079. [Google Scholar] [CrossRef]
- Assarsson, E.; Greenbaum, J.A.; Sundstrom, M.; Schaffer, L.; Hammond, J.A.; Pasquetto, V.; Oseroff, C.; Hendrickson, R.C.; Lefkowitz, E.J.; Tscharke, D.C.; et al. Kinetic analysis of a complete poxvirus transcriptome reveals an immediate-early class of genes. Proc. Natl. Acad. Sci. USA 2008, 105, 2140–2145. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.C.; Bahar, M.W.; Cooray, S.; Chen, R.A.; Whalen, D.M.; Abrescia, N.G.; Alderton, D.; Owens, R.J.; Stuart, D.I.; Smith, G.L.; et al. Vaccinia virus proteins A52 and B14 Share a Bcl-2-like fold but have evolved to inhibit NF-kappaB rather than apoptosis. PLoS Pathog. 2008, 4, e1000128. [Google Scholar] [CrossRef] [PubMed]
- Harte, M.T.; Haga, I.R.; Maloney, G.; Gray, P.; Reading, P.C.; Bartlett, N.W.; Smith, G.L.; Bowie, A.; O’Neill, L.A. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J. Exp. Med. 2003, 197, 343–351. [Google Scholar] [CrossRef]
- Chen, R.A.; Jacobs, N.; Smith, G.L. Vaccinia virus strain Western Reserve protein B14 is an intracellular virulence factor. J. Gen. Virol. 2006, 87, 1451–1458. [Google Scholar] [CrossRef]
- Smith, G.L.; Chan, Y.S.; Howard, S.T. Nucleotide sequence of 42 kbp of vaccinia virus strain WR from near the right inverted terminal repeat. J. Gen. Virol. 1991, 72 Pt 6, 1349–1376. [Google Scholar] [CrossRef]
- Gonzalez, J.M.; Esteban, M. A poxvirus Bcl-2-like gene family involved in regulation of host immune response: Sequence similarity and evolutionary history. Virol. J. 2010, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.A.; Ryzhakov, G.; Cooray, S.; Randow, F.; Smith, G.L. Inhibition of IkappaB kinase by vaccinia virus virulence factor B14. PLoS Pathog. 2008, 4, e22. [Google Scholar] [CrossRef]
- Tang, Q.; Chakraborty, S.; Xu, G. Mechanism of vaccinia viral protein B14-mediated inhibition of IkappaB kinase beta activation. J. Biol. Chem. 2018, 293, 10344–10352. [Google Scholar] [CrossRef]
- Cooray, S.; Bahar, M.W.; Abrescia, N.G.A.; McVey, C.E.; Bartlett, N.W.; Chen, R.A.; Stuart, D.I.; Grimes, J.M.; Smith, G.L. Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J. Gen. Virol. 2007, 88, 1656–1666. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, M.; Zhai, D.; Jin, C.; Aleshin, A.E.; Stec, B.; Reed, J.C.; Liddington, R.C. Vaccinia virus N1L protein resembles a B cell lymphoma-2 (Bcl-2) family protein. Protein Sci. 2007, 16, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Veyer, D.L.; Carrara, G.; Maluquer de Motes, C.; Smith, G.L. Vaccinia virus evasion of regulated cell death. Immunol. Lett. 2017, 186, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Maluquer de Motes, C.; Cooray, S.; Ren, H.; Almeida, G.M.; McGourty, K.; Bahar, M.W.; Stuart, D.I.; Grimes, J.M.; Graham, S.C.; Smith, G.L. Inhibition of apoptosis and NF-kappaB activation by vaccinia protein N1 occur via distinct binding surfaces and make different contributions to virulence. PLoS Pathog. 2011, 7, e1002430. [Google Scholar] [CrossRef]
- DiPerna, G.; Stack, J.; Bowie, A.G.; Boyd, A.; Kotwal, G.; Zhang, Z.; Arvikar, S.; Latz, E.; Fitzgerald, K.A.; Marshall, W.L. Poxvirus protein N1L targets the I-kappaB kinase complex, inhibits signaling to NF-kappaB by the tumor necrosis factor superfamily of receptors, and inhibits NF-kappaB and IRF3 signaling by toll-like receptors. J. Biol. Chem. 2004, 279, 36570–36578. [Google Scholar] [CrossRef] [PubMed]
- Kettle, S.; Alcami, A.; Khanna, A.; Ehret, R.; Jassoy, C.; Smith, G.L. Vaccinia virus serpin B13R (SPI-2) inhibits interleukin-1beta-converting enzyme and protects virus-infected cells from TNF- and Fas-mediated apoptosis, but does not prevent IL-1beta-induced fever. J. Gen. Virol. 1997, 78 Pt 3, 677–685. [Google Scholar] [CrossRef]
- Kettle, S.; Blake, N.W.; Law, K.M.; Smith, G.L. Vaccinia virus serpins B13R (SPI-2) and B22R (SPI-1) encode M(r) 38.5 and 40K, intracellular polypeptides that do not affect virus virulence in a murine intranasal model. Virology 1995, 206, 136–147. [Google Scholar] [CrossRef]
- Smith, G.L.; Howard, S.T.; Chan, Y.S. Vaccinia virus encodes a family of genes with homology to serine proteinase inhibitors. J. Gen. Virol. 1989, 70 Pt 9, 2333–2343. [Google Scholar] [CrossRef]
- Goebel, S.J.; Johnson, G.P.; Perkus, M.E.; Davis, S.W.; Winslow, J.P.; Paoletti, E. The complete DNA sequence of vaccinia virus. Virology 1990, 179, 247–266. [Google Scholar] [CrossRef]
- Chea, L.S.; Wyatt, L.S.; Gangadhara, S.; Moss, B.; Amara, R.R. Novel Modified Vaccinia Virus Ankara Vector Expressing Anti-apoptotic Gene B13R Delays Apoptosis and Enhances Humoral Responses. J. Virol. 2019, 93, e01648-18. [Google Scholar] [CrossRef]
- Antoine, G.; Scheiflinger, F.; Dorner, F.; Falkner, F.G. The complete genomic sequence of the modified vaccinia Ankara strain: Comparison with other orthopoxviruses. Virology 1998, 244, 365–396. [Google Scholar] [CrossRef] [PubMed]
- Dobbelstein, M.; Shenk, T. Protection against apoptosis by the vaccinia virus SPI-2 (B13R) gene product. J. Virol. 1996, 70, 6479–6485. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin-1 beta, interleukin-18, and the interleukin-1 beta converting enzyme. Ann. N. Y. Acad. Sci. 1998, 856, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Miao, E.A.; Ting, J.P. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity 2013, 39, 432–441. [Google Scholar] [CrossRef]
- Del Rey, A.; Verdenhalven, M.; Lorwald, A.C.; Meyer, C.; Hernangomez, M.; Randolf, A.; Roggero, E.; Konig, A.M.; Heverhagen, J.T.; Guaza, C.; et al. Brain-borne IL-1 adjusts glucoregulation and provides fuel support to astrocytes and neurons in an autocrine/paracrine manner. Mol. Psychiatry 2016, 21, 1309–1320. [Google Scholar] [CrossRef]
- Komiyama, T.; Ray, C.A.; Pickup, D.J.; Howard, A.D.; Thornberry, N.A.; Peterson, E.P.; Salvesen, G. Inhibition of interleukin-1 beta converting enzyme by the cowpox virus serpin CrmA. An example of cross-class inhibition. J. Biol. Chem. 1994, 269, 19331–19337. [Google Scholar] [CrossRef]
- Renatus, M.; Zhou, Q.; Stennicke, H.R.; Snipas, S.J.; Turk, D.; Bankston, L.A.; Liddington, R.C.; Salvesen, G.S. Crystal structure of the apoptotic suppressor CrmA in its cleaved form. Structure 2000, 8, 789–797. [Google Scholar] [CrossRef]
- Tscharke, D.C.; Reading, P.C.; Smith, G.L. Dermal infection with vaccinia virus reveals roles for virus proteins not seen using other inoculation routes. J. Gen. Virol. 2002, 83, 1977–1986. [Google Scholar] [CrossRef]
- Eaglesham, J.B.; Pan, Y.; Kupper, T.S.; Kranzusch, P.J. Publisher Correction: Viral and metazoan poxins are cGAMP-specific nucleases that restrict cGAS-STING signalling. Nature 2019, 569, E12. [Google Scholar] [CrossRef]
- Wickramasekera, N.T.; Traktman, P. Structure/Function analysis of the vaccinia virus F18 phosphoprotein, an abundant core component required for virion maturation and infectivity. J. Virol. 2010, 84, 6846–6860. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Moss, B. Vaccinia virus morphogenesis is interrupted when expression of the gene encoding an 11-kilodalton phosphorylated protein is prevented by the Escherichia coli lac repressor. J. Virol. 1991, 65, 6101–6110. [Google Scholar] [CrossRef] [PubMed]
- Meade, N.; Furey, C.; Li, H.; Verma, R.; Chai, Q.; Rollins, M.G.; DiGiuseppe, S.; Naghavi, M.H.; Walsh, D. Poxviruses Evade Cytosolic Sensing through Disruption of an mTORC1-mTORC2 Regulatory Circuit. Cell 2018, 174, 1143–1157.e1117. [Google Scholar] [CrossRef] [PubMed]
- Mansur, D.S.; Maluquer De Motes, C.; Unterholzner, L.; Sumner, R.P.; Ferguson, B.J.; Ren, H.; Strnadova, P.; Bowie, A.G.; Smith, G.L. Poxvirus Targeting of E3 Ligase β-TrCP by Molecular Mimicry: A Mechanism to Inhibit NF-κB Activation and Promote Immune Evasion and Virulence. PLoS Pathog. 2013, 9, e1003183. [Google Scholar] [CrossRef]
- Neidel, S.; Ren, H.; Torres, A.A.; Smith, G.L. NF-kappaB activation is a turn on for vaccinia virus phosphoprotein A49 to turn off NF-kappaB activation. Proc. Natl. Acad. Sci. USA 2019, 116, 5699–5704. [Google Scholar] [CrossRef]
- Bour, S.; Perrin, C.; Akari, H.; Strebel, K. The Human Immunodeficiency Virus Type 1 Vpu Protein Inhibits NF-κB Activation by Interfering with βTrCP-mediated Degradation of IκB. J. Biol. Chem. 2001, 276, 15920–15928. [Google Scholar] [CrossRef]
- Albarnaz, J.D.; Ren, H.; Torres, A.A.; Shmeleva, E.V.; Melo, C.A.; Bannister, A.J.; Brember, M.P.; Chung, B.Y.W.; Smith, G.L. Molecular mimicry of NF-κB by vaccinia virus protein enables selective inhibition of antiviral responses. Nat. Microbiol. 2022, 7, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Yang, X.-D.; Zhou, M.-M.; Ozato, K.; Chen, L.-F. Brd4 Coactivates Transcriptional Activation of NF-κB via Specific Binding to Acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Patel, D. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 1999, 18, 5061–5072. [Google Scholar] [CrossRef] [Green Version]
- Marzio, G.; Tyagi, M.; Gutierrez, M.I.; Giacca, M. HIV-1 tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc. Natl. Acad. Sci. USA 1998, 95, 13519–13524. [Google Scholar] [CrossRef]
- O’Connor, M.J.; Zimmermann, H.; Nielsen, S.; Bernard, H.U.; Kouzarides, T. Characterization of an E1A-CBP interaction defines a novel transcriptional adapter motif (TRAM) in CBP/p300. J. Virol. 1999, 73, 3574–3581. [Google Scholar] [CrossRef]
- Gillard, S.; Spehner, D.; Drillien, R.; Kirn, A. Localization and sequence of a vaccinia virus gene required for multiplication in human cells. Proc. Natl. Acad. Sci. USA 1986, 83, 5573–5577. [Google Scholar] [CrossRef] [PubMed]
- Sivan, G.; Ormanoglu, P.; Buehler, E.C.; Martin, S.E.; Moss, B. Identification of Restriction Factors by Human Genome-Wide RNA Interference Screening of Viral Host Range Mutants Exemplified by Discovery of SAMD9 and WDR6 as Inhibitors of the Vaccinia Virus K1L-C7L- Mutant. mBio 2015, 6, e01122. [Google Scholar] [CrossRef] [PubMed]
- Perkus, M.E.; Goebel, S.J.; Davis, S.W.; Johnson, G.P.; Limbach, K.; Norton, E.K.; Paoletti, E. Vaccinia virus host range genes. Virology 1990, 179, 276–286. [Google Scholar] [CrossRef]
- Bravo Cruz, A.G.; Shisler, J.L. Vaccinia virus K1 ankyrin repeat protein inhibits NF-κB activation by preventing RelA acetylation. J. Gen. Virol. 2016, 97, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Bruno, D.P.; Martens, C.A.; Porcella, S.F.; Moss, B. Simultaneous high-resolution analysis of vaccinia virus and host cell transcriptomes by deep RNA sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 11513–11518. [Google Scholar] [CrossRef]
- Beard, P.M.; Froggatt, G.C.; Smith, G.L. Vaccinia virus kelch protein A55 is a 64 kDa intracellular factor that affects virus-induced cytopathic effect and the outcome of infection in a murine intradermal model. J. Gen. Virol. 2006, 87, 1521–1529. [Google Scholar] [CrossRef]
- Pintard, L.; Willems, A.; Peter, M. Cullin-based ubiquitin ligases: Cul3–BTB complexes join the family. EMBO J. 2004, 23, 1681–1687. [Google Scholar] [CrossRef]
- Pallett, M.A.; Ren, H.; Zhang, R.Y.; Scutts, S.R.; Gonzalez, L.; Zhu, Z.; Maluquer de Motes, C.; Smith, G.L. Vaccinia Virus BBK E3 Ligase Adaptor A55 Targets Importin-Dependent NF-kappaB Activation and Inhibits CD8(+) T-Cell Memory. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Liang, P.; Zhang, H.; Wang, G.; Li, S.; Cong, S.; Luo, Y.; Zhang, B. KPNB1, XPO7 and IPO8 mediate the translocation ofNF-kappaB/p65 into the nucleus. Traffic 2013, 14, 1132–1143. [Google Scholar] [CrossRef]
- Ember, S.W.J.; Ren, H.; Ferguson, B.J.; Smith, G.L. Vaccinia virus protein C4 inhibits NF-κB activation and promotes virus virulence. J. Gen. Virol. 2012, 93, 2098–2108. [Google Scholar] [CrossRef]
- Smith, K.A.; Stallard, V.; Roos, J.M.; Hart, C.; Cormier, N.; Cohen, L.K.; Roberts, B.E.; Payne, L.G. Host range selection of vaccinia recombinants containing insertions of foreign genes into non-coding sequences. Vaccine 1993, 11, 43–53. [Google Scholar] [CrossRef]
- Hotokezaka, H.; Sakai, E.; Kanaoka, K.; Saito, K.; Matsuo, K.; Kitaura, H.; Yoshida, N.; Nakayama, K. U0126 and PD98059, specific inhibitors of MEK, accelerate differentiation of RAW264.7 cells into osteoclast-like cells. J. Biol. Chem. 2002, 277, 47366–47372. [Google Scholar] [CrossRef] [PubMed]
- Gedey, R.; Jin, X.L.; Hinthong, O.; Shisler, J.L. Poxviral regulation of the host NF-kappaB response: The vaccinia virus M2L protein inhibits induction of NF-kappaB activation via an ERK2 pathway in virus-infected human embryonic kidney cells. J. Virol. 2006, 80, 8676–8685. [Google Scholar] [CrossRef] [PubMed]
- Hinthong, O.; Jin, X.L.; Shisler, J.L. Characterization of wild-type and mutant vaccinia virus M2L proteins' abilities to localize to the endoplasmic reticulum and to inhibit NF-kappaB activation during infection. Virology 2008, 373, 248–262. [Google Scholar] [CrossRef]
- Moss, B.; Shisler, J.L. Immunology 101 at poxvirus U: Immune evasion genes. Semin. Immunol. 2001, 13, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Ray, C.A.; Black, R.A.; Kronheim, S.R.; Greenstreet, T.A.; Sleath, P.R.; Salvesen, G.S.; Pickup, D.J. Viral inhibition of inflammation: Cowpox virus encodes an inhibitor of the interleukin-1 beta converting enzyme. Cell 1992, 69, 597–604. [Google Scholar] [CrossRef]
- Petit, F.; Bertagnoli, S.; Gelfi, J.; Fassy, F.; Boucraut-Baralon, C.; Milon, A. Characterization of a myxoma virus-encoded serpin-like protein with activity against interleukin-1 beta-converting enzyme. J. Virol. 1996, 70, 5860–5866. [Google Scholar] [CrossRef]
- Dorfleutner, A.; Talbott, S.J.; Bryan, N.B.; Funya, K.N.; Rellick, S.L.; Reed, J.C.; Shi, X.; Rojanasakul, Y.; Flynn, D.C.; Stehlik, C. A Shope Fibroma virus PYRIN-only protein modulates the host immune response. Virus Genes 2007, 35, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Diel, D.G.; Luo, S.; Delhon, G.; Peng, Y.; Flores, E.F.; Rock, D.L. A nuclear inhibitor of NF-kappaB encoded by a poxvirus. J. Virol. 2011, 85, 264–275. [Google Scholar] [CrossRef]
- Ning, Z.; Zheng, Z.; Hao, W.; Duan, C.; Li, W.; Wang, Y.; Li, M.; Luo, S. The N terminus of orf virus-encoded protein 002 inhibits acetylation of NF-kappaB p65 by preventing Ser(276) phosphorylation. PLoS ONE 2013, 8, e58854. [Google Scholar] [CrossRef]
- Nichols, D.B.; Shisler, J.L. Poxvirus MC160 protein utilizes multiple mechanisms to inhibit NF-kappaB activation mediated via components of the tumor necrosis factor receptor 1 signal transduction pathway. J. Virol. 2009, 83, 3162–3174. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Inhibitors targeting receptors mediating NF-κB activation | |||||
| WR Gene | Copenhagen Gene | Expression | Localization | Mechanism of NF-κB Inhibition | Reference |
| 032 | K1L | Early | Cytoplasmic | Limits dsRNA production to prevent PKR stimulation | [70,89,92] |
| 034 | K3L | Early | Cytoplasmic | eIF-2α mimic | [82,83,84] |
| 059 | E3L | Early/Late | Cytoplasmic | Inhibits PKR activation as an RNA-binding protein | [73,75,76,77] |
| 196 | B15R | Early | Extracellular | Inteleukin-1β-binding protein | [100,102,103] |
| 013 | C12L | Early/Late | Extracellular | Inteleukin-18-binding protein | [93,94,95] |
| Inhibitors targeting NF-κB signaling intermediates | |||||
| WR Gene | Copenhagen Gene | Expression | Localization | Mechanism of NF-κB Inhibition | Reference |
| 028 | N1L | Early/Late | Cytoplasmic | Inhibits IKK complex members, facilitating NF-κB activation | [126,127] |
| 039 | K7R | Early | Cytoplasmic | TRAF6 and IRAK2 interaction inhibiting NF-κB activation | [104,106] |
| 056 | F17R | Late | Cytoplasmic | mTOR dysregulation leading to cGAS degradation | [144] |
| 172 | A46R | Early/Late | Cytoplasmic | Targets TIR-domain-containing adaptor proteins (e.g., MyD88, Mal) | [107,109,112,113,114] |
| 178 | A52R | Early/Late | Cytoplasmic | TRAF6 and IRAK2 interaction inhibiting NF-κB activation; Targets host TIR domain-containing proteins (e.g., MyD88) | [109,117] |
| 184 | B2R | Early | Cytoplasmic | 2′3′-cGAMP nuclease inhibiting cGAS-STING signaling | [141] |
| 195 | B13R | Early | Cytoplasmic | Blocks proteolytic activity of ICE/Caspase-1 | [128,134] |
| 196 | B14R | Early | Cytoplasmic | Prevents IKKβ trans-auto-phosphorylation; Sterically hinders IKKβ-IKK complex formation | [121,122] |
| Inhibitors directly targeting NF-κB complex activation/activity | |||||
| WR Gene | Copenhagen Gene | Expression | Localization | Mechanism of NF-κB Inhibition | Reference |
| 024 | C4L | Early | Cytoplasmic | Prevents nuclear translocation of p65/RelA | [162] |
| 031 | M2L | Early | Cytoplasmic | ERK1/2 antagonist | [165,166] |
| 032 | K1L | Early | Cytoplasmic Nuclear | IκBα degradation inhibitor; Prevents acetylation of NF-κB subunit p65/RelA | [156] |
| 053 | F14L | Late | Nuclear | Inhibits acetylation of NF-κB subunit p65/RelA | [148] |
| 175 | A49R | Early/Late | Cytoplasmic | Interacts with β-TRCP to prevent ubiquitination of IκBα | [149] |
| 180 | A55R | Early | Cytoplasmic | Inhibits importin α-dependent nuclear translocation of NF-κB | [159,160,161] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reus, J.B.; Rex, E.A.; Gammon, D.B. How to Inhibit Nuclear Factor-Kappa B Signaling: Lessons from Poxviruses. Pathogens 2022, 11, 1061. https://doi.org/10.3390/pathogens11091061
Reus JB, Rex EA, Gammon DB. How to Inhibit Nuclear Factor-Kappa B Signaling: Lessons from Poxviruses. Pathogens. 2022; 11(9):1061. https://doi.org/10.3390/pathogens11091061
Chicago/Turabian StyleReus, Joshua B., Emily A. Rex, and Don B. Gammon. 2022. "How to Inhibit Nuclear Factor-Kappa B Signaling: Lessons from Poxviruses" Pathogens 11, no. 9: 1061. https://doi.org/10.3390/pathogens11091061