
Phage ImmunoPrecipitation Sequencing (PhIP-Seq): The Promise of High Throughput Serology


Abstract
:Simple Summary
Abstract
1. Introduction
2. Phage Display for Serology prior to PhIP-Seq
3. PhIP-Seq Technology
3.1. Phage Library Construction
3.2. Phage Library Propagation
3.3. Phage Immunoprecipitation
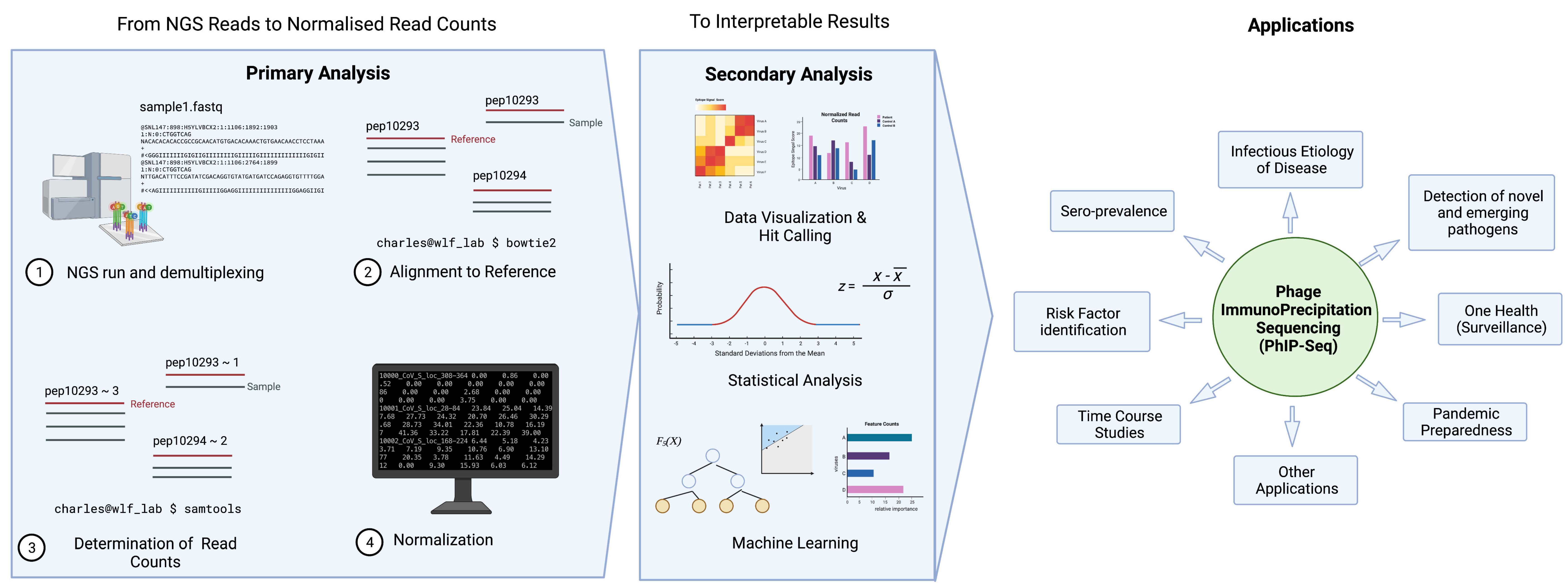
3.4. Data Analysis Overview

3.5. Determining the Hits
3.6. Other Analysis Strategies (Machine Learning, AVARDA, Novel Pipelines)
3.7. Programming Language and Skills Needed
4. Improving PhIP-Seq
5. Applications of PhIP-Seq in Sero-Epidemiology
5.1. Seroprevalence Studies
5.2. Risk Factor Analysis and Association Studies
5.3. Vaccinology and Response to Vaccines
5.4. Time Course Studies
5.5. Infectious Etiology of Disease Studies
5.6. Novel and Emerging Pathogens
5.7. Applications to One Health and Pandemic Preparedness
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Petherick, A. Developing antibody tests for SARS-CoV-2. Lancet 2020, 395, 1101–1102. [Google Scholar] [CrossRef]
- Jones, K.; Savulescu, A.F.; Brombacher, F.; Hadebe, S. Immunoglobulin M in Health and Diseases: How Far Have We Come and What Next? Front. Immunol. 2020, 11, 595535. [Google Scholar] [CrossRef]
- Deeks, J.J.; Dinnes, J.; Takwoingi, Y.; Davenport, C.; Spijker, R.; Taylor-Phillips, S.; Adriano, A.; Beese, S.; Dretzke, J.; Ferrante di Ruffano, L.; et al. Antibody tests for identification of current and past infection with SARS-CoV-2. Cochrane Database Syst. Rev. 2020, 6, Cd013652. [Google Scholar] [CrossRef]
- Freihorst, J.; Ogra, P.L. Mucosal immunity and viral infections. Ann. Med. 2001, 33, 172–177. [Google Scholar] [CrossRef]
- Mandal, S.; Das, H.; Deo, S.; Arinaminpathy, N. Combining serology with case-detection, to allow the easing of restrictions against SARS-CoV-2: A modelling-based study in India. Sci. Rep. 2021, 11, 1835. [Google Scholar] [CrossRef]
- Dowlatshahi, S.; Shabani, E.; Abdekhodaie, M.J. Serological assays and host antibody detection in coronavirus-related disease diagnosis. Arch. Virol. 2021, 166, 715–731. [Google Scholar] [CrossRef]
- Davies, D.H.; Liang, X.; Hernandez, J.E.; Randall, A.; Hirst, S.; Mu, Y.; Romero, K.M.; Nguyen, T.T.; Kalantari-Dehaghi, M.; Crotty, S.; et al. Profiling the humoral immune response to infection by using proteome microarrays: High-throughput vaccine and diagnostic antigen discovery. Proc. Natl. Acad. Sci. USA 2005, 102, 547–552. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.; Fornace, K.; Wu, L.; Arnold, B.F.; Priest, J.W.; Martin, D.L.; Chang, M.A.; Cook, J.; Stresman, G.; Drakeley, C. Determining seropositivity—A review of approaches to define population seroprevalence when using multiplex bead assays to assess burden of tropical diseases. PLoS Negl. Trop. Dis. 2021, 15, e0009457. [Google Scholar] [CrossRef]
- Larman, H.B.; Zhao, Z.; Laserson, U.; Li, M.Z.; Ciccia, A.; Gakidis, M.A.; Church, G.M.; Kesari, S.; Leproust, E.M.; Solimini, N.L.; et al. Autoantigen discovery with a synthetic human peptidome. Nat. Biotechnol. 2011, 29, 535–541. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.P. Phage Display: Simple Evolution in a Petri Dish (Nobel Lecture). Angew. Chem. Int. Ed. 2019, 58, 14428–14437. [Google Scholar] [CrossRef] [Green Version]
- Alfaleh, M.A.; Alsaab, H.O.; Mahmoud, A.B.; Alkayyal, A.A.; Jones, M.L.; Mahler, S.M.; Hashem, A.M. Phage Display Derived Monoclonal Antibodies: From Bench to Bedside. Front. Immunol. 2020, 11, 1986. [Google Scholar] [CrossRef] [PubMed]
- Midoro-Horiuti, T.; Goldblum, R.M. Epitope mapping with random phage display library. Methods Mol. Biol. 2014, 1131, 477–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hust, M.; Lim, T.S. Phage Display; Springer: New York, NY, USA, 2018. [Google Scholar]
- Krumpe, L.R.; Mori, T. The Use of Phage-Displayed Peptide Libraries to Develop Tumor-Targeting Drugs. Int. J. Pept. Res. Ther. 2006, 12, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Caberoy, N.B.; Alvarado, G.; Davis, J.L.; Feuer, W.J.; Li, W. Identification of Hnrph3 as an autoantigen for acute anterior uveitis. Clin. Immunol. 2011, 138, 60–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yu, J.; Sreekumar, A.; Varambally, S.; Shen, R.; Giacherio, D.; Mehra, R.; Montie, J.E.; Pienta, K.J.; Sanda, M.G.; et al. Autoantibody Signatures in Prostate Cancer. N. Engl. J. Med. 2005, 353, 1224–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faix, P.H.; Burg, M.A.; Gonzales, M.; Ravey, E.P.; Baird, A.; Larocca, D. Phage display of cDNA libraries: Enrichment of cDNA expression using open reading frame selection. BioTechniques 2004, 36, 1018–1029. [Google Scholar] [CrossRef]
- Georgieva, Y.; Konthur, Z. Design and screening of M13 phage display cDNA libraries. Molecules 2011, 16, 1667–1681. [Google Scholar] [CrossRef]
- Shrock, E.; Fujimura, E.; Kula, T.; Timms, R.T.; Lee, I.H.; Leng, Y.; Robinson, M.L.; Sie, B.M.; Li, M.Z.; Chen, Y.; et al. Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity. Science 2020, 370, eabd4250. [Google Scholar] [CrossRef]
- Xu, G.J.; Kula, T.; Xu, Q.; Li, M.Z.; Vernon, S.D.; Ndung’u, T.; Ruxrungtham, K.; Sanchez, J.; Brander, C.; Chung, R.T.; et al. Viral immunology. Comprehensive serological profiling of human populations using a synthetic human virome. Science 2015, 348, aaa0698. [Google Scholar] [CrossRef] [Green Version]
- Schubert, R.D.; Hawes, I.A.; Ramachandran, P.S.; Ramesh, A.; Crawford, E.D.; Pak, J.E.; Wu, W.; Cheung, C.K.; O’Donovan, B.D.; Tato, C.M.; et al. Pan-viral serology implicates enteroviruses in acute flaccid myelitis. Nat. Med. 2019, 25, 1748–1752. [Google Scholar] [CrossRef]
- Vogl, T.; Klompus, S.; Leviatan, S.; Kalka, I.N.; Weinberger, A.; Wijmenga, C.; Fu, J.; Zhernakova, A.; Weersma, R.K.; Segal, E. Population-wide diversity and stability of serum antibody epitope repertoires against human microbiota. Nat. Med. 2021, 27, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Mohan, D.; Wansley, D.L.; Sie, B.M.; Noon, M.S.; Baer, A.N.; Laserson, U.; Larman, H.B. PhIP-Seq characterization of serum antibodies using oligonucleotide-encoded peptidomes. Nat. Protoc. 2018, 13, 1958–1978. [Google Scholar] [CrossRef] [PubMed]
- Mina, M.J.; Kula, T.; Leng, Y.; Li, M.; de Vries, R.D.; Knip, M.; Siljander, H.; Rewers, M.; Choy, D.F.; Wilson, M.S.; et al. Measles virus infection diminishes preexisting antibodies that offer protection from other pathogens. Science 2019, 366, 599–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, M.R.; Rahman, M.; Khan, T.; Saeed, A.; Sundararaju, S.; Flores, A.; Hawken, P.; Rawat, A.; Elkum, N.; Hussain, K.; et al. Virome-wide serological profiling reveals association of herpesviruses with obesity. Sci. Rep. 2021, 11, 2562. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Caberoy, N.B. New perspective for phage display as an efficient and versatile technology of functional proteomics. Appl. Microbiol. Biotechnol. 2010, 85, 909–919. [Google Scholar] [CrossRef] [Green Version]
- Irving, A.T.; Rozario, P.; Kong, P.-S.; Luko, K.; Gorman, J.J.; Hastie, M.L.; Chia, W.N.; Mani, S.; Lee, B.P.Y.H.; Smith, G.J.D.; et al. Robust dengue virus infection in bat cells and limited innate immune responses coupled with positive serology from bats in IndoMalaya and Australasia. Cell. Mol. Life Sci. 2020, 77, 1607–1622. [Google Scholar] [CrossRef]
- Ravichandran, S.; Hahn, M.; Belaunzarán-Zamudio, P.F.; Ramos-Castañeda, J.; Nájera-Cancino, G.; Caballero-Sosa, S.; Navarro-Fuentes, K.R.; Ruiz-Palacios, G.; Golding, H.; Beigel, J.H.; et al. Differential human antibody repertoires following Zika infection and the implications for serodiagnostics and disease outcome. Nat. Commun. 2019, 10, 1943. [Google Scholar] [CrossRef]
- Monaco, D.R.; Sie, B.M.; Nirschl, T.R.; Knight, A.C.; Sampson, H.A.; Nowak-Wegrzyn, A.; Wood, R.A.; Hamilton, R.G.; Frischmeyer-Guerrerio, P.A.; Larman, H.B. Profiling serum antibodies with a pan allergen phage library identifies key wheat allergy epitopes. Nat. Commun. 2021, 12, 379. [Google Scholar] [CrossRef]
- Stoddard, C.I.; Galloway, J.; Chu, H.Y.; Shipley, M.M.; Sung, K.; Itell, H.L.; Wolf, C.R.; Logue, J.K.; Magedson, A.; Garrett, M.E.; et al. Epitope profiling reveals binding signatures of SARS-CoV-2 immune response in natural infection and cross-reactivity with endemic human CoVs. Cell Rep. 2021, 35, 109164. [Google Scholar] [CrossRef]
- Eshleman, S.H.; Laeyendecker, O.; Kammers, K.; Chen, A.; Sivay, M.V.; Kottapalli, S.; Sie, B.M.; Yuan, T.; Monaco, D.R.; Mohan, D.; et al. Comprehensive Profiling of HIV Antibody Evolution. Cell Rep. 2019, 27, 1422–1433.e4. [Google Scholar] [CrossRef] [Green Version]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tang, W.; Budhu, A.; Forgues, M.; Hernandez, M.O.; Candia, J.; Kim, Y.; Bowman, E.D.; Ambs, S.; Zhao, Y.; et al. A Viral Exposure Signature Defines Early Onset of Hepatocellular Carcinoma. Cell 2020, 182, 317–328.e10. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.P.; Larman, H.B.; Lee, M.H.; Whitehead, S.S.; Kowalak, J.; Toro, C.; Lau, C.C.; Kim, J.; Johnson, K.R.; Reoma, L.B.; et al. Chronic Dengue Virus Panencephalitis in a Patient with Progressive Dementia with Extrapyramidal Features. Ann. Neurol. 2019, 86, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Leon, K.E.; Schubert, R.D.; Casas-Alba, D.; Hawes, I.A.; Ramachandran, P.S.; Ramesh, A.; Pak, J.E.; Wu, W.; Cheung, C.K.; Crawford, E.D.; et al. Genomic and serologic characterization of enterovirus A71 brainstem encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e703. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Monaco, D.R.; Kottapalli, S.V.; Breitwieser, F.P.; Anderson, D.E.; Wijaya, L.; Tan, K.; Chia, W.N.; Kammers, K.; Caturegli, P.; Waugh, K.; et al. Deconvoluting virome-wide antibody epitope reactivity profiles. eBioMedicine 2021, 75, 103747. [Google Scholar] [CrossRef]
- Morales, P.; Jiménez, M.A. Design and structural characterisation of monomeric water-soluble α-helix and β-hairpin peptides: State-of-the-art. Arch. Biochem. Biophys. 2019, 661, 149–167. [Google Scholar] [CrossRef]
- Rey, F.A.; Lok, S.M. Common Features of Enveloped Viruses and Implications for Immunogen Design for Next-Generation Vaccines. Cell 2018, 172, 1319–1334. [Google Scholar] [CrossRef]
- Román-Meléndez, G.D.; Monaco, D.R.; Montagne, J.M.; Quizon, R.S.; Konig, M.F.; Astatke, M.; Darrah, E.; Larman, H.B. Citrullination of a phage-displayed human peptidome library reveals the fine specificities of rheumatoid arthritis-associated autoantibodies. eBioMedicine 2021, 71, 103506. [Google Scholar] [CrossRef]
- Garrett, M.E.; Galloway, J.G.; Wolf, C.; Logue, J.K.; Franko, N.; Chu, H.Y.; Matsen, F.A., IV; Overbaugh, J.M. Comprehensive characterization of the antibody responses to SARS-CoV-2 Spike protein finds additional vaccine-induced epitopes beyond those for mild infection. eLife 2022, 11, e73490. [Google Scholar] [CrossRef]
- Smith, C.C.; Olsen, K.S.; Gentry, K.M.; Sambade, M.; Beck, W.; Garness, J.; Entwistle, S.; Willis, C.; Vensko, S.; Woods, A.; et al. Landscape and selection of vaccine epitopes in SARS-CoV-2. Genome Med. 2021, 13, 101. [Google Scholar] [CrossRef]
- Bender Ignacio, R.A.; Dasgupta, S.; Stevens-Ayers, T.; Kula, T.; Hill, J.A.; Lee, S.J.; Mielcarek, M.; Duerr, A.; Elledge, S.J.; Boeckh, M. Comprehensive viromewide antibody responses by systematic epitope scanning after hematopoietic cell transplantation. Blood 2019, 134, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Morgenlander, W.R.; Henson, S.N.; Monaco, D.R.; Chen, A.; Littlefield, K.; Bloch, E.M.; Fujimura, E.; Ruczinski, I.; Crowley, A.R.; Natarajan, H.; et al. Antibody responses to endemic coronaviruses modulate COVID-19 convalescent plasma functionality. J. Clin. Investig. 2021, 131, e146927. [Google Scholar] [CrossRef] [PubMed]
- Klompus, S.; Leviatan, S.; Vogl, T.; Mazor, R.D.; Kalka, I.N.; Stoler-Barak, L.; Nathan, N.; Peres, A.; Moss, L.; Godneva, A.; et al. Cross-reactive antibodies against human coronaviruses and the animal coronavirome suggest diagnostics for future zoonotic spillovers. Sci. Immunol. 2021, 6, eabe9950. [Google Scholar] [CrossRef] [PubMed]
- Amuasi, J.H.; Lucas, T.; Horton, R.; Winkler, A.S. Reconnecting for our future: The Lancet One Health Commission. Lancet 2020, 395, 1469–1471. [Google Scholar] [CrossRef]
- The European Union One Health 2019 Zoonoses Report. EFSA J. 2021, 19, e06406. [CrossRef]
- Otu, A.; Effa, E.; Meseko, C.; Cadmus, S.; Ochu, C.; Athingo, R.; Namisango, E.; Ogoina, D.; Okonofua, F.; Ebenso, B. Africa needs to prioritize One Health approaches that focus on the environment, animal health and human health. Nat. Med. 2021, 27, 943–946. [Google Scholar] [CrossRef]
- Delai, R.R.; Freitas, A.R.; Kmetiuk, L.B.; Merigueti, Y.; Ferreira, I.B.; Lescano, S.A.Z.; Gonzáles, W.H.R.; Brandão, A.P.D.; de Barros-Filho, I.R.; Pettan-Brewer, C.; et al. One Health approach on human seroprevalence of anti-Toxocara antibodies, Toxocara spp. eggs in dogs and sand samples between seashore mainland and island areas of southern Brazil. One Health 2021, 13, 100353. [Google Scholar] [CrossRef]
- Gilbert, A.T.; Fooks, A.R.; Hayman, D.T.; Horton, D.L.; Muller, T.; Plowright, R.; Peel, A.J.; Bowen, R.; Wood, J.L.; Mills, J.; et al. Deciphering serology to understand the ecology of infectious diseases in wildlife. Ecohealth 2013, 10, 298–313. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Shrock, E.L.; Li, M.Z.; Spergel, J.M.; Nadeau, K.C.; Pongracic, J.A.; Umetsu, D.T.; Rachid, R.; MacGinnitie, A.J.; Phipatanakul, W.; et al. High-resolution epitope mapping by AllerScan reveals relationships between IgE and IgG repertoires during peanut oral immunotherapy. Cell Rep. Med. 2021, 2, 100410. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Assay | Solid Phase | Antigen | Detection | Multi-Plexability |
|---|---|---|---|---|
| ELISA | Plastic plate | Whole proteins or subdomains of interest | Enzyme-tagged antibody | Limited |
| Western Blot | Nitrocellulose Membrane | Denatured proteins | Enzyme-tagged antibody | Limited |
| IFA | Cells | Cells expressing protein of interest | Fluorescent-tagged antibody | Limited |
| Luminex | Beads | Whole proteins or subdomains of interest | Fluorescent-tagged antibody | Medium |
| PhIP-Seq | T7 Bacteriophage | Peptide tiles | NGS | High |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiu, C.K.; Zhu, F.; Wang, L.-F.; de Alwis, R. Phage ImmunoPrecipitation Sequencing (PhIP-Seq): The Promise of High Throughput Serology. Pathogens 2022, 11, 568. https://doi.org/10.3390/pathogens11050568
Tiu CK, Zhu F, Wang L-F, de Alwis R. Phage ImmunoPrecipitation Sequencing (PhIP-Seq): The Promise of High Throughput Serology. Pathogens. 2022; 11(5):568. https://doi.org/10.3390/pathogens11050568
Chicago/Turabian StyleTiu, Charles Kevin, Feng Zhu, Lin-Fa Wang, and Ruklanthi de Alwis. 2022. "Phage ImmunoPrecipitation Sequencing (PhIP-Seq): The Promise of High Throughput Serology" Pathogens 11, no. 5: 568. https://doi.org/10.3390/pathogens11050568