
Generation and Validation of Monoclonal Antibodies Suitable for Detecting and Monitoring Parvovirus Infections


 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Antigen Design and Screening Approach to Generate mAbs Specific for NS1 from Rodent PVs, Human CuV, and b19v
2.2. Identification of Hybridoma Producing mAbs for NS1 Detection
2.2.1. H-1PV NS1
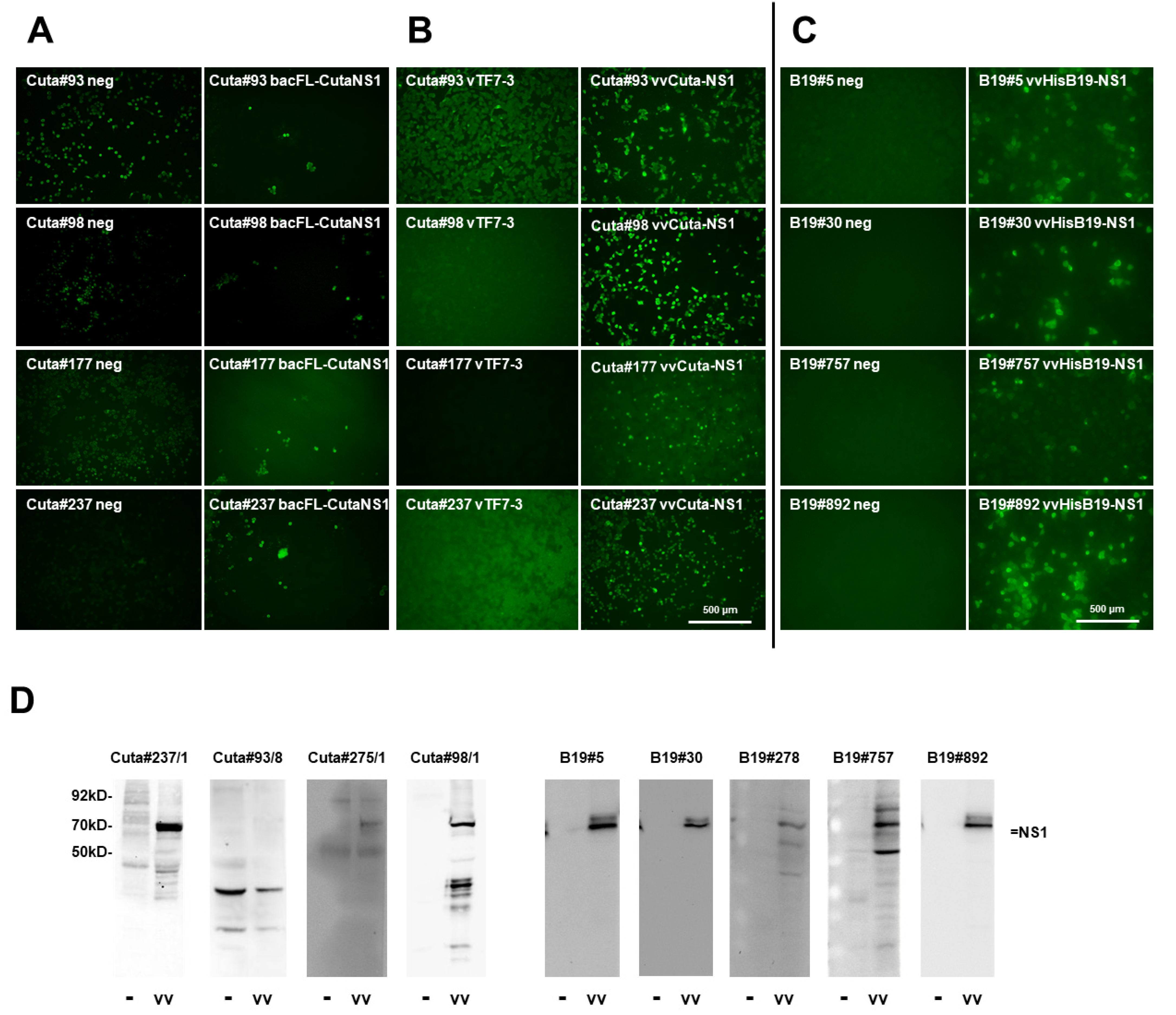
2.2.2. CuV NS1
2.2.3. B19V NS1
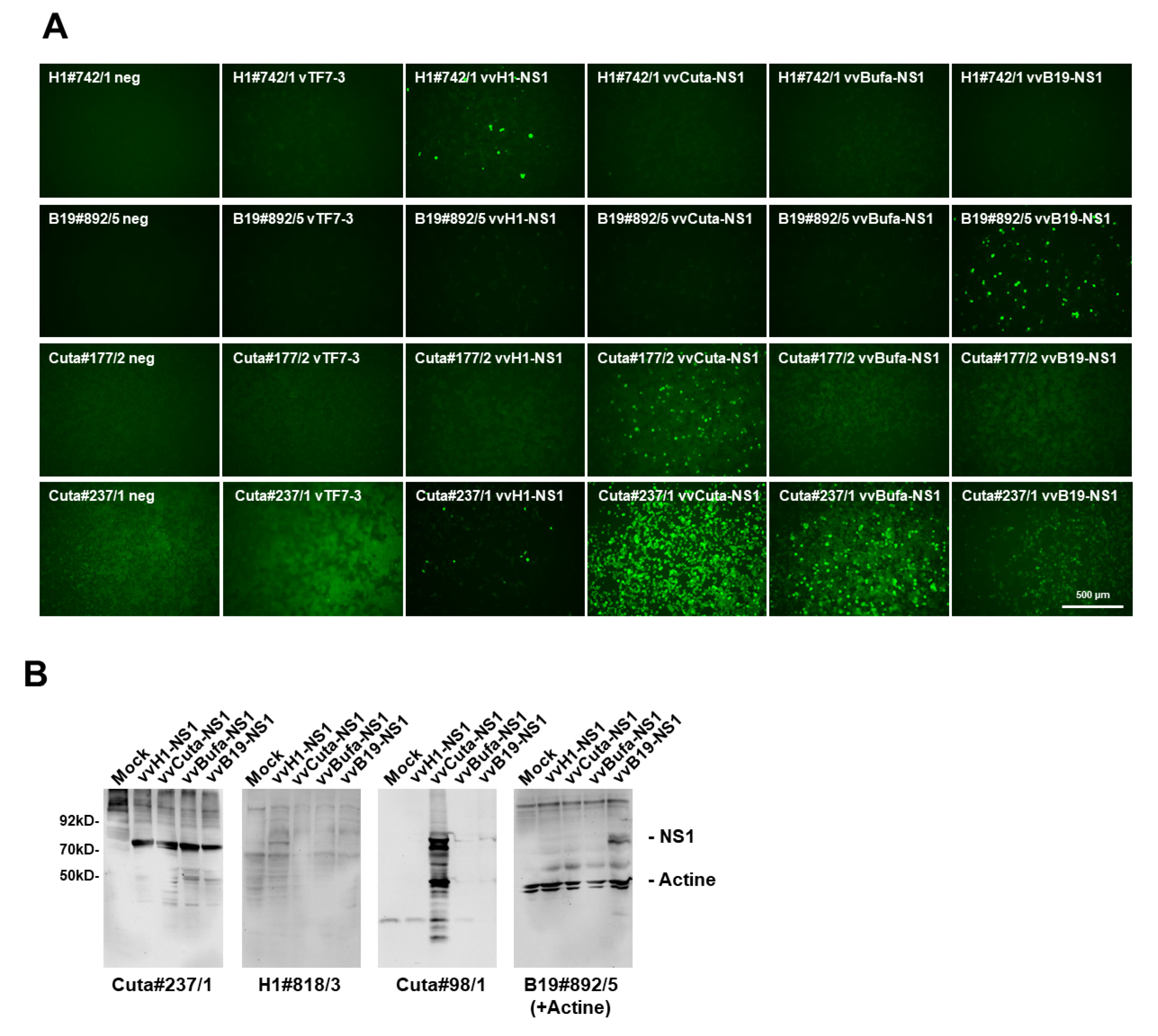
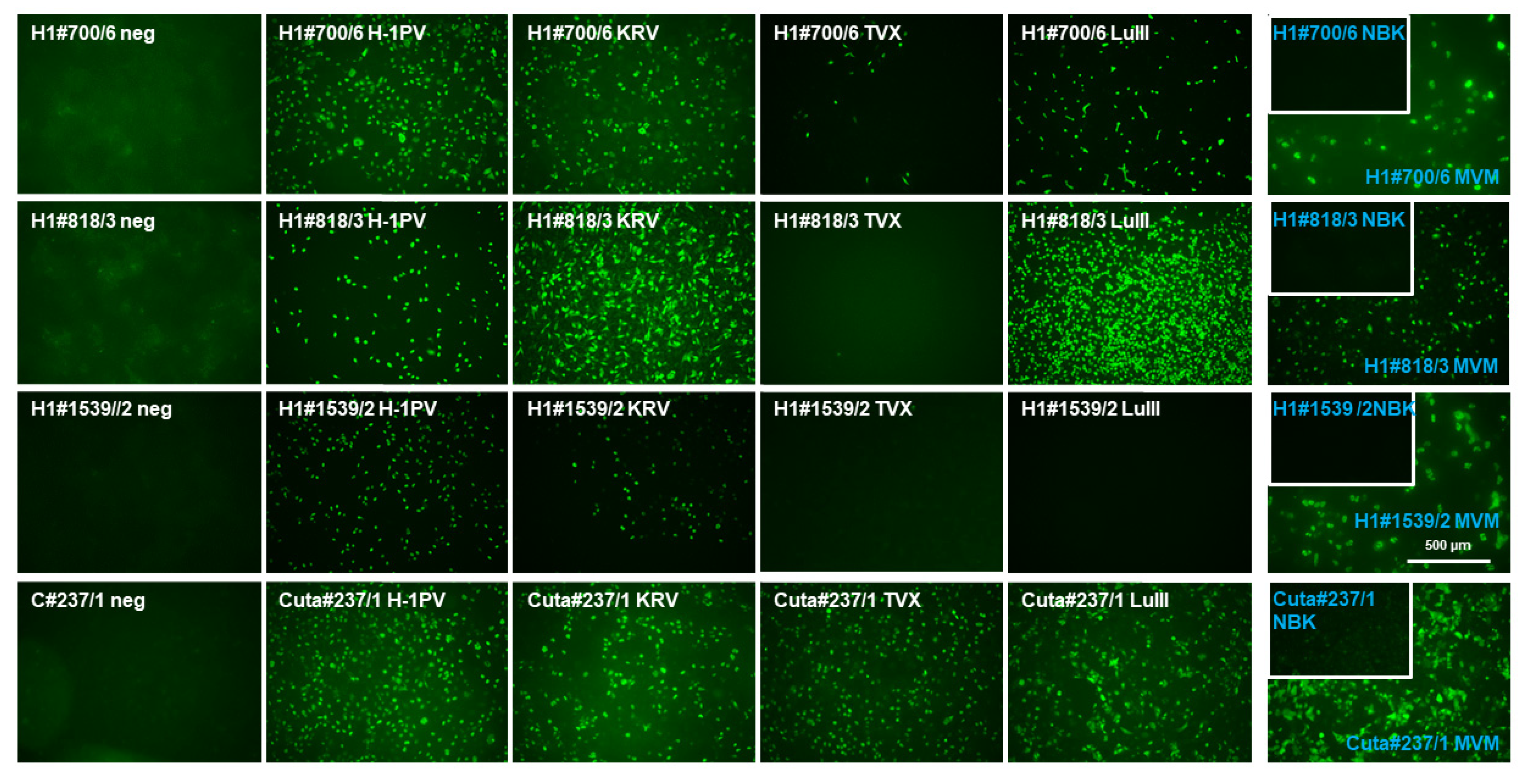
2.3. Assessment of Antibody Cross-Reactivity between Rodent and Human Parvoviruses
2.4. Monoclonal Antibodies Detecting the C-Terminal Part of NS1
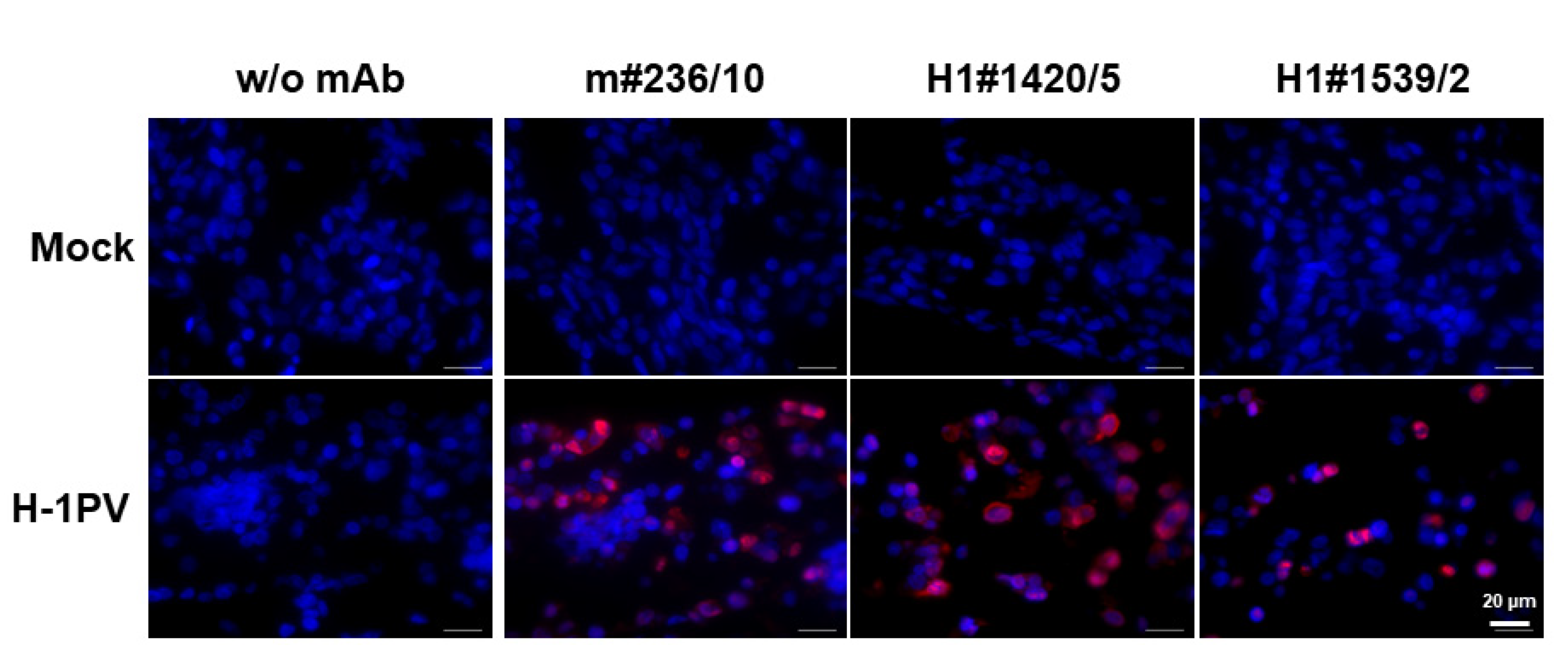
2.5. Monoclonal Antibodies Detecting H-1PV in Paraffin-Embedded Samples
3. Discussion
4. Material and Methods
4.1. Animal Ethics Statement
4.2. DNA Constructs
4.3. Reagents
4.4. Cells and Viruses
4.5. Bacterial Expression and Purification of Immunogens
4.6. Immunization, Cell Fusion, and the Isolation of Monoclonal Antibodies
4.7. Immunofluorescence Microscopy
4.8. Western Blot Analysis
4.9. Paraffin Embedment of Cultured Cells and Detection of Target Proteins
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cotmore, S.F.; Tattersall, P. The autonomously replicating parvoviruses of vertebrates. Adv. Virus Res. 1987, 33, 91–174. [Google Scholar] [CrossRef] [PubMed]
- Bashir, T.; Horlein, R.; Rommelaere, J.; Willwand, K. Cyclin A activates the DNA polymerase delta -dependent elongation machinery in vitro: A parvovirus DNA replication model. Proc. Natl. Acad. Sci. USA 2000, 97, 5522–5527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallauer, C.; Kronauer, G.; Siegl, G. Parvoiruses as contaminants of permanent human cell lines. I. Virus isolation from 1960-1970. Arch. Gesamte Virusforsch 1971, 35, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Streck, A.F.; Truyen, U. Porcine Parvovirus. Curr. Issues Mol. Biol. 2020, 37, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truyen, U.; Parrish, C.R. Feline panleukopenia virus: Its interesting evolution and current problems in immunoprophylaxis against a serious pathogen. Vet. Microbiol. 2013, 165, 29–32. [Google Scholar] [CrossRef]
- Soderlund-Venermo, M. Emerging Human Parvoviruses: The Rocky Road to Fame. Annu. Rev. Virol. 2019, 6, 71–91. [Google Scholar] [CrossRef]
- Qiu, J.; Soderlund-Venermo, M.; Young, N.S. Human Parvoviruses. Clin. Microbiol. Rev. 2017, 30, 43–113. [Google Scholar] [CrossRef] [Green Version]
- Adamson-Small, L.A.; Ignatovich, I.V.; Laemmerhirt, M.G.; Hobbs, J.A. Persistent parvovirus B19 infection in non-erythroid tissues: Possible role in the inflammatory and disease process. Virus Res. 2014, 190, 8–16. [Google Scholar] [CrossRef]
- Phan, T.G.; Dreno, B.; da Costa, A.C.; Li, L.; Orlandi, P.; Deng, X.; Kapusinszky, B.; Siqueira, J.; Knol, A.C.; Halary, F.; et al. A new protoparvovirus in human fecal samples and cutaneous T cell lymphomas (mycosis fungoides). Virology 2016, 496, 299–305. [Google Scholar] [CrossRef]
- Mollerup, S.; Fridholm, H.; Vinner, L.; Kjartansdottir, K.R.; Friis-Nielsen, J.; Asplund, M.; Herrera, J.A.; Steiniche, T.; Mourier, T.; Brunak, S.; et al. Cutavirus in Cutaneous Malignant Melanoma. Emerg. Infect. Dis. 2017, 23, 363–365. [Google Scholar] [CrossRef] [Green Version]
- Zadori, Z.; Szelei, J.; Tijssen, P. SAT: A late NS protein of porcine parvovirus. J. Virol. 2005, 79, 13129–13138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashir, T.; Rommelaere, J.; Cziepluch, C. In vivo accumulation of cyclin A and cellular replication factors in autonomous parvovirus minute virus of mice-associated replication bodies. J. Virol. 2001, 75, 4394–4398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuesch, J.P.; Rommelaere, J. NS1 interaction with CKII alpha: Novel protein complex mediating parvovirus-induced cytotoxicity. J. Virol. 2006, 80, 4729–4739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar, S.; Rommelaere, J.; Nuesch, J.P. PKCeta/Rdx-driven phosphorylation of PDK1: A novel mechanism promoting cancer cell survival and permissiveness for parvovirus-induced lysis. PLoS Pathog. 2015, 11, e1004703. [Google Scholar] [CrossRef] [Green Version]
- Bar, S.; Rommelaere, J.; Nuesch, J.P. Vesicular transport of progeny parvovirus particles through ER and Golgi regulates maturation and cytolysis. PLoS Pathog. 2013, 9, e1003605. [Google Scholar] [CrossRef] [Green Version]
- Nuesch, J.P.; Lachmann, S.; Rommelaere, J. Selective alterations of the host cell architecture upon infection with parvovirus minute virus of mice. Virology 2005, 331, 159–174. [Google Scholar] [CrossRef] [Green Version]
- Nuesch, J.P.; Lacroix, J.; Marchini, A.; Rommelaere, J. Molecular pathways: Rodent parvoviruses--mechanisms of oncolysis and prospects for clinical cancer treatment. Clin. Cancer Res. 2012, 18, 3516–3523. [Google Scholar] [CrossRef] [Green Version]
- Cotmore, S.F.; Tattersall, P. Alternate splicing in a parvoviral nonstructural gene links a common amino-terminal sequence to downstream domains which confer radically different localization and turnover characteristics. Virology 1990, 177, 477–487. [Google Scholar] [CrossRef]
- Brown, K.E.; Young, N.S.; Liu, J.M. Molecular, cellular and clinical aspects of parvovirus B19 infection. Crit. Rev. Oncol Hematol. 1994, 16, 1–31. [Google Scholar] [CrossRef]
- Gigler, A.; Dorsch, S.; Hemauer, A.; Williams, C.; Kim, S.; Young, N.S.; Zolla-Pazner, S.; Wolf, H.; Gorny, M.K.; Modrow, S. Generation of neutralizing human monoclonal antibodies against parvovirus B19 proteins. J. Virol. 1999, 73, 1974–1979. [Google Scholar] [CrossRef] [Green Version]
- Yeung, D.E.; Brown, G.W.; Tam, P.; Russnak, R.H.; Wilson, G.; Clark-Lewis, I.; Astell, C.R. Monoclonal antibodies to the major nonstructural nuclear protein of minute virus of mice. Virology 1991, 181, 35–45. [Google Scholar] [CrossRef]
- Kerr, J.R. Pathogenesis of parvovirus B19 infection: Host gene variability, and possible means and effects of virus persistence. J. Vet. Med. B Infect. Dis. Vet. Public Health 2005, 52, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. Organization of nonstructural genes of the autonomous parvovirus minute virus of mice. J. Virol. 1986, 58, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockhaus, K.; Plaza, S.; Pintel, D.J.; Rommelaere, J.; Salome, N. Nonstructural proteins NS2 of minute virus of mice associate in vivo with 14-3-3 protein family members. J. Virol. 1996, 70, 7527–7534. [Google Scholar] [CrossRef] [Green Version]
- Nüesch, J. Regulation of non-structural protein functions by differ 445 ential synthesis, modifications and trafficking. In Parvoviruses; Kerr, C.S., Jr., Bloom, M.E., Linden, R.M., Parrish, C.R., Eds.; Edward 447 Arnold Ltd.: London, UK, 2005; pp. 275–290. [Google Scholar]
- Bodendorf, U.; Cziepluch, C.; Jauniaux, J.C.; Rommelaere, J.; Salome, N. Nuclear export factor CRM1 interacts with nonstructural proteins NS2 from parvovirus minute virus of mice. J. Virol. 1999, 73, 7769–7779. [Google Scholar] [CrossRef] [Green Version]
- Christensen, J.; Cotmore, S.F.; Tattersall, P. Minute virus of mice transcriptional activator protein NS1 binds directly to the transactivation region of the viral P38 promoter in a strictly ATP-dependent manner. J. Virol. 1995, 69, 5422–5430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuesch, J.P.; Tattersall, P. Nuclear targeting of the parvoviral replicator molecule NS1: Evidence for self-association prior to nuclear transport. Virology 1993, 196, 637–651. [Google Scholar] [CrossRef]
- Corbau, R.; Salom, N.; Rommelaere, J.; Nuesch, J.P. Phosphorylation of the viral nonstructural protein NS1 during MVMp infection of A9 cells. Virology 1999, 259, 402–415. [Google Scholar] [CrossRef]
- Nuesch, J.P.; Corbau, R.; Tattersall, P.; Rommelaere, J. Biochemical activities of minute virus of mice nonstructural protein NS1 are modulated In vitro by the phosphorylation state of the polypeptide. J. Virol. 1998, 72, 8002–8012. [Google Scholar] [CrossRef] [Green Version]
- Nuesch, J.P.; Dettwiler, S.; Corbau, R.; Rommelaere, J. Replicative functions of minute virus of mice NS1 protein are regulated in vitro by phosphorylation through protein kinase C. J. Virol. 1998, 72, 9966–9977. [Google Scholar] [CrossRef] [Green Version]
- Nuesch, J.P.; Cotmore, S.F.; Tattersall, P. Expression of functional parvoviral NS1 from recombinant vaccinia virus: Effects of mutations in the nucleotide-binding motif. Virology 1992, 191, 406–416. [Google Scholar] [CrossRef]
- Nuesch, J.P.; Rommelaere, J. Tumor suppressing properties of rodent parvovirus NS1 proteins and their derivatives. Adv. Exp. Med. Biol. 2014, 818, 99–124. [Google Scholar] [CrossRef] [PubMed]
- Rhode, S.L., 3rd; Paradiso, P.R. Parvovirus genome: Nucleotide sequence of H-1 and mapping of its genes by hybrid-arrested translation. J. Virol. 1983, 45, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diffoot, N.; Chen, K.C.; Bates, R.C.; Lederman, M. The complete nucleotide sequence of parvovirus LuIII and localization of a unique sequence possibly responsible for its encapsidation pattern. Virology 1993, 192, 339–345. [Google Scholar] [CrossRef]
- Merchlinsky, M.J.; Tattersall, P.J.; Leary, J.J.; Cotmore, S.F.; Gardiner, E.M.; Ward, D.C. Construction of an infectious molecular clone of the autonomous parvovirus minute virus of mice. J. Virol. 1983, 47, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Vollmers, E.M.; D’Abramo, A., Jr.; Cotmore, S.F.; Tattersall, P. Genome sequence of tumor virus x, a member of the genus protoparvovirus in the family parvoviridae. Genome Announc. 2014, 2, e00758-14. [Google Scholar] [CrossRef] [Green Version]
- Faisst, S.; Faisst, S.R.; Dupressoir, T.; Plaza, S.; Pujol, A.; Jauniaux, J.C.; Rhode, S.L.; Rommelaere, J. Isolation of a fully infectious variant of parvovirus H-1 supplanting the standard strain in human cells. J. Virol. 1995, 69, 4538–4543. [Google Scholar] [CrossRef] [Green Version]
- Nüesch, J.; Thomas, N.; Plotzky, C.; Rommelaere, J. Modified Rodent Parvovirus Capable of Propagating and Spreading through Human Gliomas. U.S. Patent 9,029,117, 12 May 2015. [Google Scholar]
- Bar, S.; Daeffler, L.; Rommelaere, J.; Nuesch, J.P. Vesicular egress of non-enveloped lytic parvoviruses depends on gelsolin functioning. PLoS Pathog. 2008, 4, e1000126. [Google Scholar] [CrossRef]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.O.; Schoning, T.; Husing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef] [Green Version]
- Angelova, A.L.; Barf, M.; Geletneky, K.; Unterberg, A.; Rommelaere, J. Immunotherapeutic Potential of Oncolytic H-1 Parvovirus: Hints of Glioblastoma Microenvironment Conversion towards Immunogenicity. Viruses 2017, 9, 382. [Google Scholar] [CrossRef] [Green Version]
- Phan, T.G.; Vo, N.P.; Bonkoungou, I.J.; Kapoor, A.; Barro, N.; O’Ryan, M.; Kapusinszky, B.; Wang, C.; Delwart, E. Acute diarrhea in West African children: Diverse enteric viruses and a novel parvovirus genus. J. Virol. 2012, 86, 11024–11030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, T.G.; Sdiri-Loulizi, K.; Aouni, M.; Ambert-Balay, K.; Pothier, P.; Deng, X.; Delwart, E. New parvovirus in child with unexplained diarrhea, Tunisia. Emerg. Infect. Dis. 2014, 20, 1911–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daeffler, L.; Horlein, R.; Rommelaere, J.; Nuesch, J.P. Modulation of minute virus of mice cytotoxic activities through site-directed mutagenesis within the NS coding region. J. Virol. 2003, 77, 12466–12478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geletneky, K.; Huesing, J.; Rommelaere, J.; Schlehofer, J.R.; Leuchs, B.; Dahm, M.; Krebs, O.; von Knebel Doeberitz, M.; Huber, B.; Hajda, J. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of Parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer 2012, 12, 99. [Google Scholar] [CrossRef] [Green Version]
- Fuerst, T.R.; Niles, E.G.; Studier, F.W.; Moss, B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. USA 1986, 83, 8122–8126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H-1PV NS1 Screening | |||||
| IF-positive | WB-positive | ||||
| 253 | 225 | ||||
| Excellent 1 | Intermediate 2 | Weak 3 | Excellent | Intermediate | Poor |
| 90 | 101 | 62 | 32 | 50 | 94 |
| CuV NS1 Screening | |||||
| IF-positive | WB-positive | ||||
| 16 | 3 | ||||
| B19V NS1 Screening | |||||
| IF-positive | WB-positive | ||||
| 10 | 5 | ||||
| Name | neg | vTF7-3 | H1 | Cuta | Bufa | B19 | KRV | TVX | LuIII | MVM | Hg#1 | Hg#13 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H-1PV-NS1-specific | ||||||||||||
| H1#742/1 | - | - | +++ | - | - | - | +++ | - | - | +++ | +++ | - |
| H1#1420/5 | - | - | +++ | - | - | - | +++ | - | - | +++ | +++ | - |
| H1#700/6 | - | ++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ |
| H1#818/3 | - | - | +++ | - | - | - | +++ | - | +++ | +++ | +++ | +++ |
| H1#1539/2 | - | n.d. | +++ | n.d. | n.d. | n.d. | +++ | - | - | +++ | +++ | - |
| H1#705 | - | n.d. | +++ | n.d. | n.d. | n.d. | +++ | +++ | ++ | - | - | +++ |
| H1#721 | - | n.d. | +++ | n.d. | n.d. | n.d. | + | - | ++ | ++ | +++ | +++ |
| H1#737 | - | n.d. | +++ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | +++ | +++ |
| H1#756 | - | n.d. | +++ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | +++ | +++ |
| H1#853 | - | n.d. | +++ | n.d. | n.d. | n.d. | ++ | - | + | ++ | +++ | +++ |
| H1#867 | - | n.d. | +++ | n.d. | n.d. | n.d. | +++ | - | +++ | +++ | +++ | +++ |
| H1#888/1 | - | n.d. | +++ | n.d. | n.d. | n.d. | +++ | - | +++ | +++ | +++ | +++ |
| H1#892/5 | - | n.d. | +++ | n.d. | n.d. | n.d. | +++ | - | - | +++ | +++ | - |
| H1#905 | - | n.d. | +++ | n.d. | n.d. | n.d. | +++ | - | - | +++ | +++ | - |
| H1#918 | - | n.d. | +++ | n.d. | n.d. | n.d. | +++ | +++ | +++ | +++ | +++ | +++ |
| H1#1252 | - | n.d. | +++ | n.d. | n.d. | n.d. | +++ | - | - | ++ | +++ | - |
| H1#1320 | - | n.d. | +++ | n.d. | n.d. | n.d. | +++ | - | ++ | +++ | +++ | +++ |
| H1#1428/5 | - | n.d. | +++ | n.d. | n.d. | n.d. | +++ | - | - | (+) | +++ | - |
| H1#1509/3 | - | n.d. | +++ | n.d. | n.d. | n.d. | +++ | +++ | +++ | +++ | +++ | +++ |
| H1#1581/3 | - | n.d. | +++ | n.d. | n.d. | n.d. | +++ | +++ | - | +++ | +++ | +++ |
| B19-NS1-specific | ||||||||||||
| B19#892/5 | - | - | - | - | - | +++ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| B19#757/10 | - | - | - | - | - | +++ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| B19#5/12/3 | - | - | (+) | (+) | (+) | +++ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| B19#278/3 | - | +++ | +++ | +++ | +++ | +++ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Cuta-NS1-specific | ||||||||||||
| C#177/2 | - | - | - | +++ | - | - | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| C#98/1 | - | - | - | +++ | + | - | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| C#93/8 | - | + | + | +++ | +++ | +++ | +++ | +++ | +++ | +++ | n.d. | n.d. |
| C#237/1 | - | - | +++ | +++ | +++ | ++ | +++ | +++ | +++ | +++ | +++ | +++ |
| Others | ||||||||||||
| pNS1H | - | - | +++ | +++ | +++ | +++ | +++ | +++ | +++ | +++ | n.d. | n.d. |
| pNS1-MK/SP8 | - | - | +++ | ++ | - | - | +++ | +++ | +++ | +++ | n.d. | n.d. |
| NS1-3D9 | - | - | +++ | - | - | - | +++ | - | +++ | +++ | n.d. | n.d. |
| m#236/10 | - | - | +++ | - | - | - | +++ | - | +++ | +++ | +++ | - |
| Forward Primer | Backward Primer | |
|---|---|---|
| Immunogens | ||
| H1-NS1Heli | ggatccgcatggctagcaccagaacctgtagaatctttgc | gcggccgcttaaataactggtgttggttcaatctgtttgc |
| H1-NS1TA | ggatccgcatggcttgttactgtgctaaatggggcaaagt | gcggccgcttagtccaaggtcagctcctcgttgaagtcgc |
| B19-NS1Heli | gaaatttcctatgtgctgcttaaacaaacaaatgggaaaaag | gcggccgctattcgttggttgtcattatgactggtgttgg |
| B19-NS1TA | gtttaaacctatgggcgcctggaacactgaaaccccgcgctc | gcggccgcttactcataatctacaaagctttgcaatcc |
| Cuta-NS1Im | gaaatttcctatgtgctgcttaaacaaacaaatgggaaaaag | aagcttgcggccgcctaatagctggcattcacatccgttccggatatc. |
| Recombinant Viruses | ||
| FL-H1-NS1 | gtttaaacatggctgactacaaggacgacgatggctggaaacgcttactc | gcggccgcttactcataatctacaaagctttgcaatcc |
| Y-CutaNS1 | gtttaaacatgagaccagagatcacgtggttgatggctctctcagcaaagagatg | aagcttgcggccgcctaatagctggcattcacatccgttccggatatc |
| FL-CutaNS1 | gtttaaacatggctgactacaaggacgacgatggctctcagcaaagagatg | aagcttgcggccgcctaatagctggcattcacatccgttccggatatc |
| FL-B19NS1 | gtttaaacatggctgactacaaggacgacgatggagctatttagaggggtg | gcggccgcttactcataatctacaaagctttgcaatcc |
| HisB19NS1 | gtttaaacatgcactaccaccactaccac atggagctatttagaggggtg | gcggccgcttactcataatctacaaagctttgcaatcc |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tessmer, C.; Plotzky, C.; Fees, J.; Welsch, H.; Eudenbach, R.; Faber, M.; Simón, A.; Angelova, A.; Rommelaere, J.; Hofmann, I.; et al. Generation and Validation of Monoclonal Antibodies Suitable for Detecting and Monitoring Parvovirus Infections. Pathogens 2022, 11, 208. https://doi.org/10.3390/pathogens11020208
Tessmer C, Plotzky C, Fees J, Welsch H, Eudenbach R, Faber M, Simón A, Angelova A, Rommelaere J, Hofmann I, et al. Generation and Validation of Monoclonal Antibodies Suitable for Detecting and Monitoring Parvovirus Infections. Pathogens. 2022; 11(2):208. https://doi.org/10.3390/pathogens11020208
Chicago/Turabian StyleTessmer, Claudia, Claudia Plotzky, Jana Fees, Hendrik Welsch, Rebecca Eudenbach, Martin Faber, Alicia Simón, Assia Angelova, Jean Rommelaere, Ilse Hofmann, and et al. 2022. "Generation and Validation of Monoclonal Antibodies Suitable for Detecting and Monitoring Parvovirus Infections" Pathogens 11, no. 2: 208. https://doi.org/10.3390/pathogens11020208