
HIV-1 Accessory Proteins Impart a Modest Interferon Response and Upregulate Cell Cycle-Related Genes in Macrophages

 ,
,  , , , and
, , , and 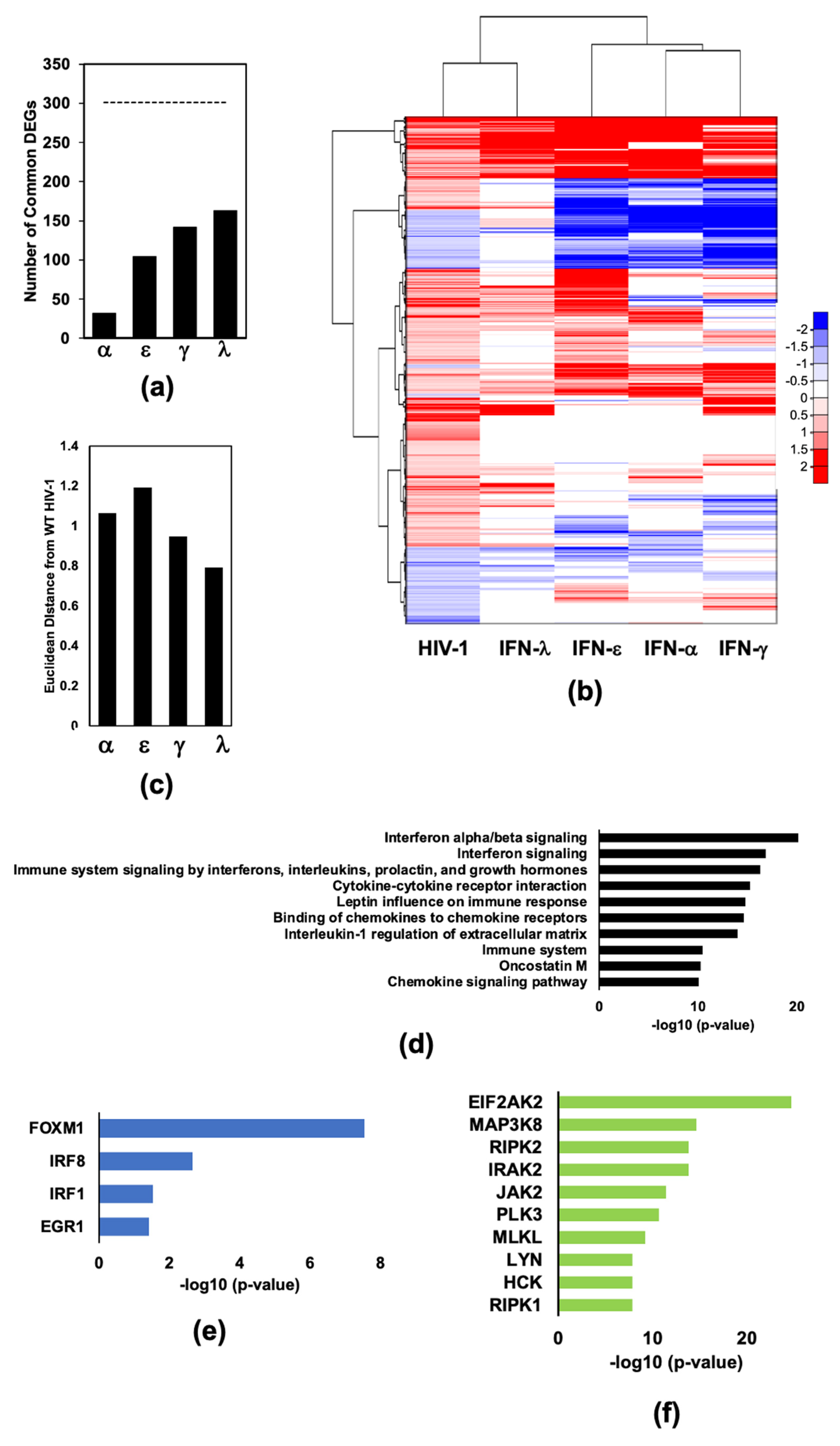{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
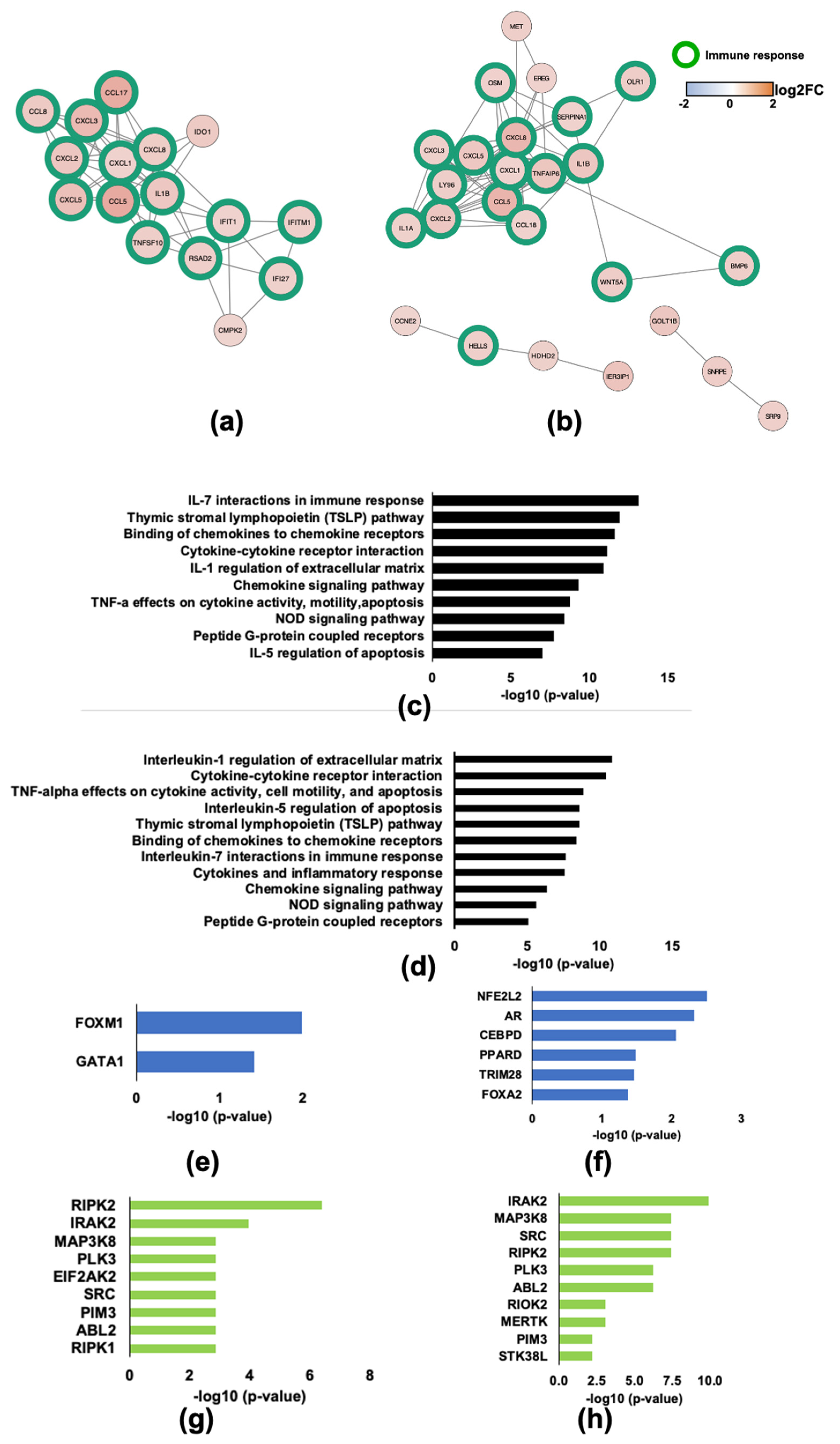
2.1. Infection of Monocyte Derived Macrophages with HIV-1Bal Induces an IFN-Like Response
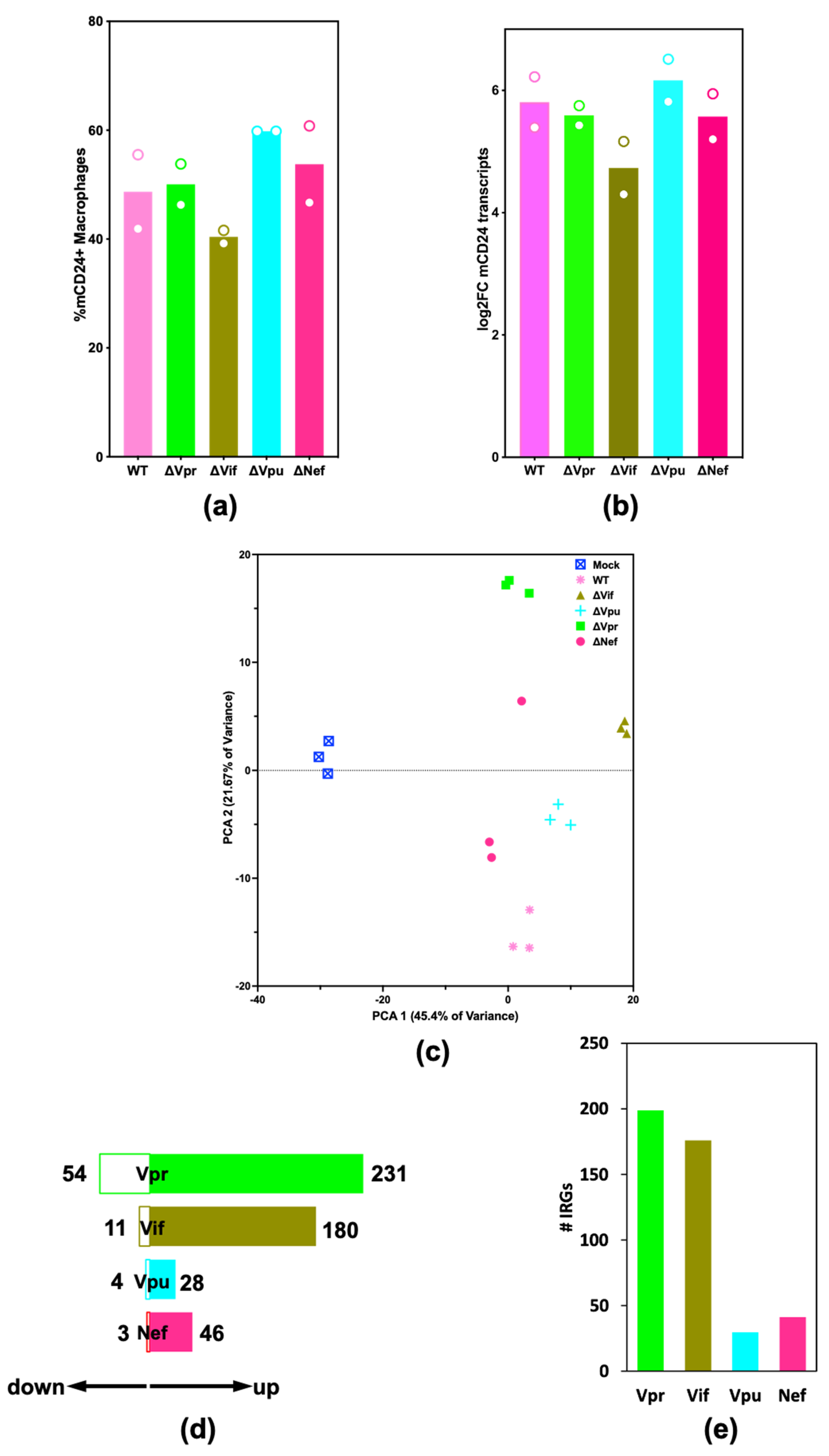
2.2. HIV-1 Vif and Vpr Induce Significant Transcriptome Changes in Macrophages Infected with HIV-1
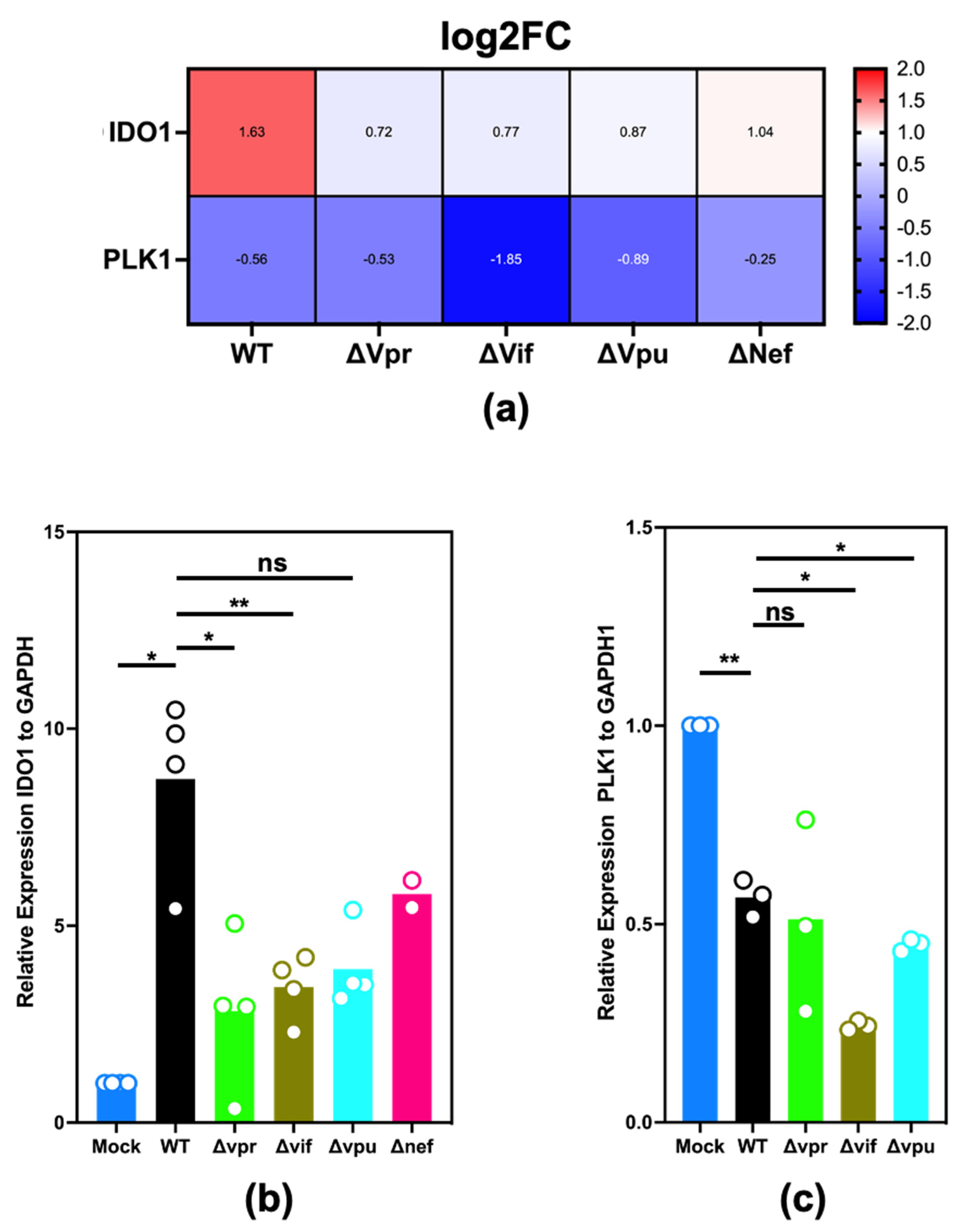
2.3. Validation of Differential Expression of IDO1 and PLK1
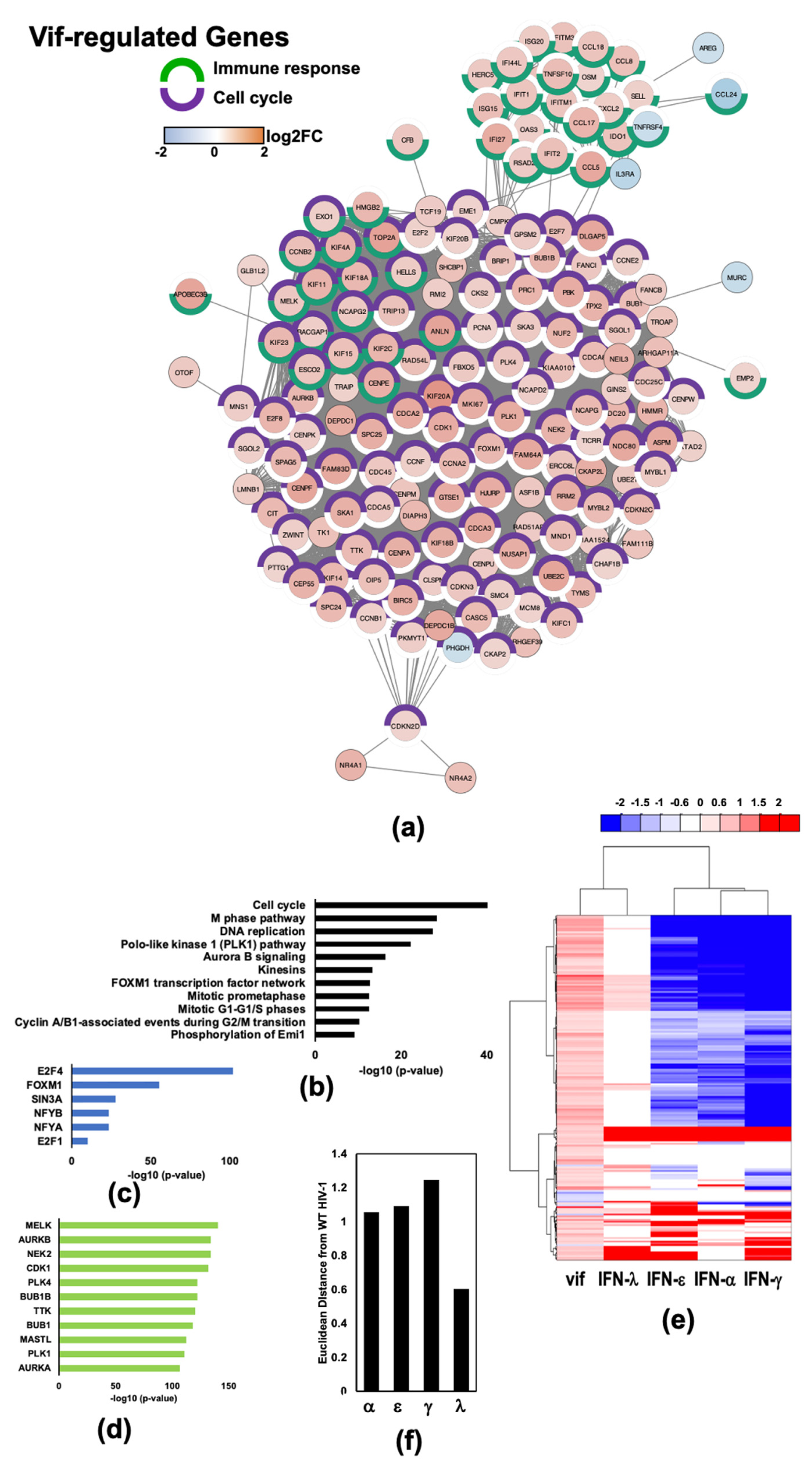
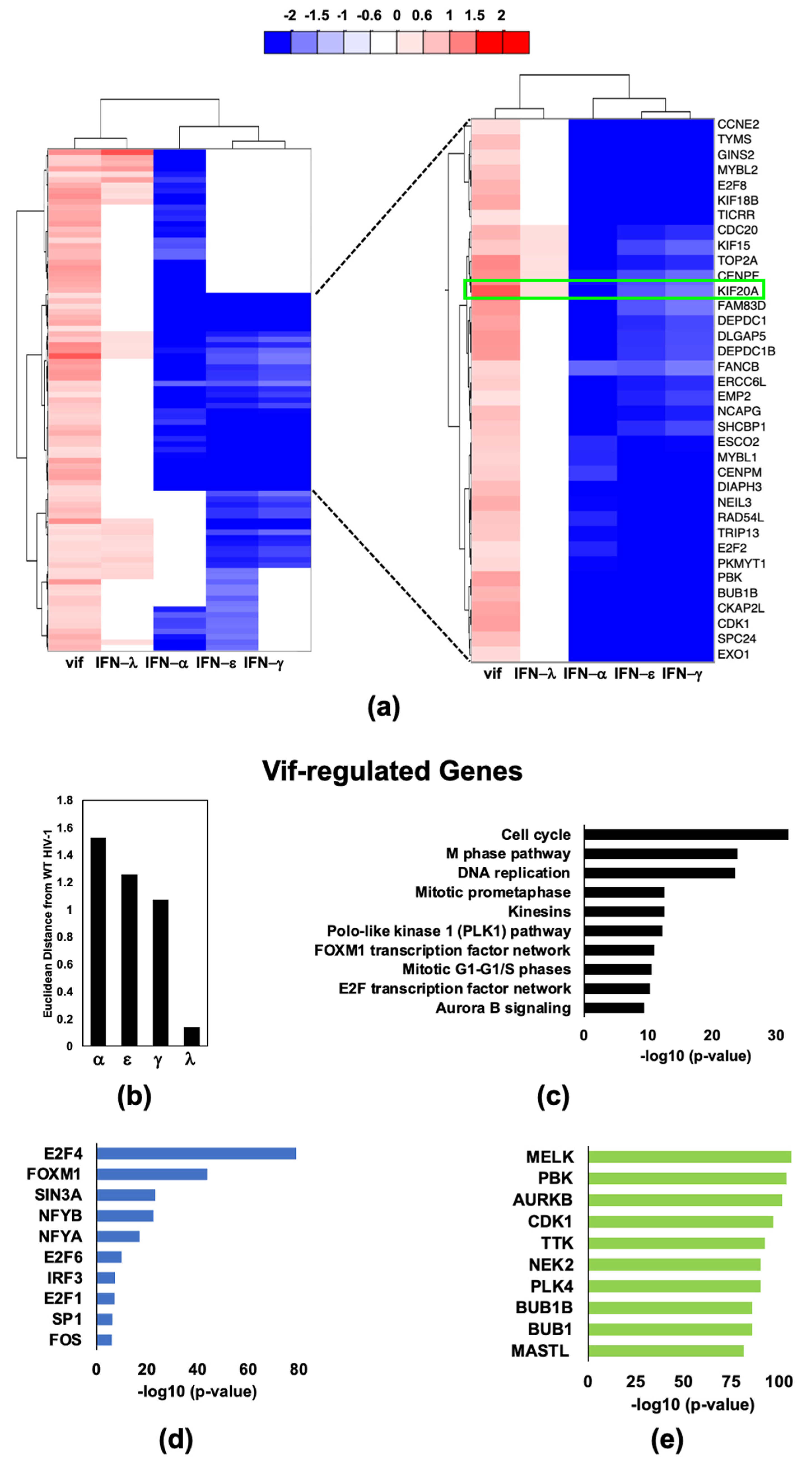
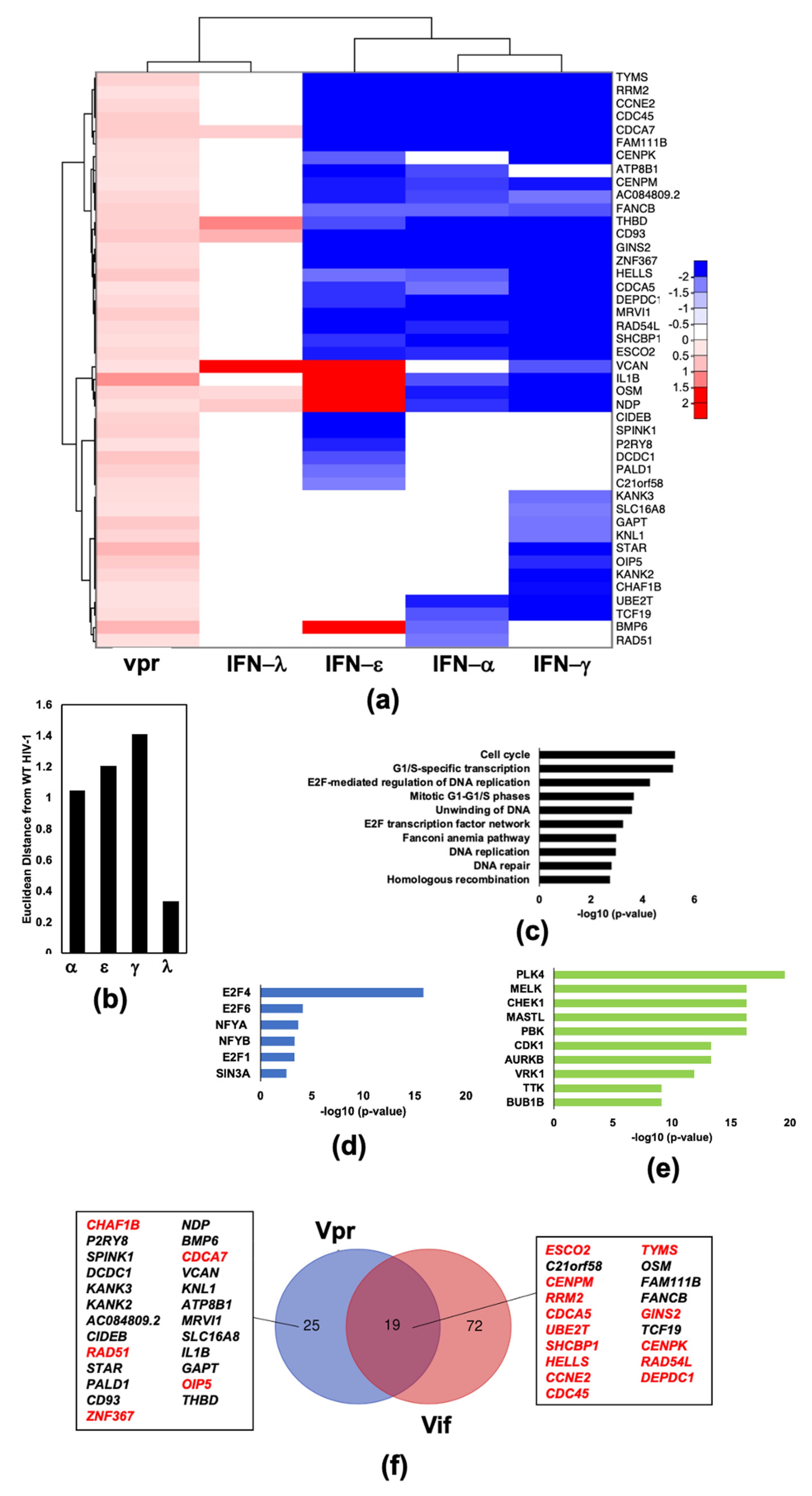
2.4. HIV-1 Vif Induces Transcriptome Changes from Genes Related to Mitosis and the Microtubule Network in Infected Macrophages
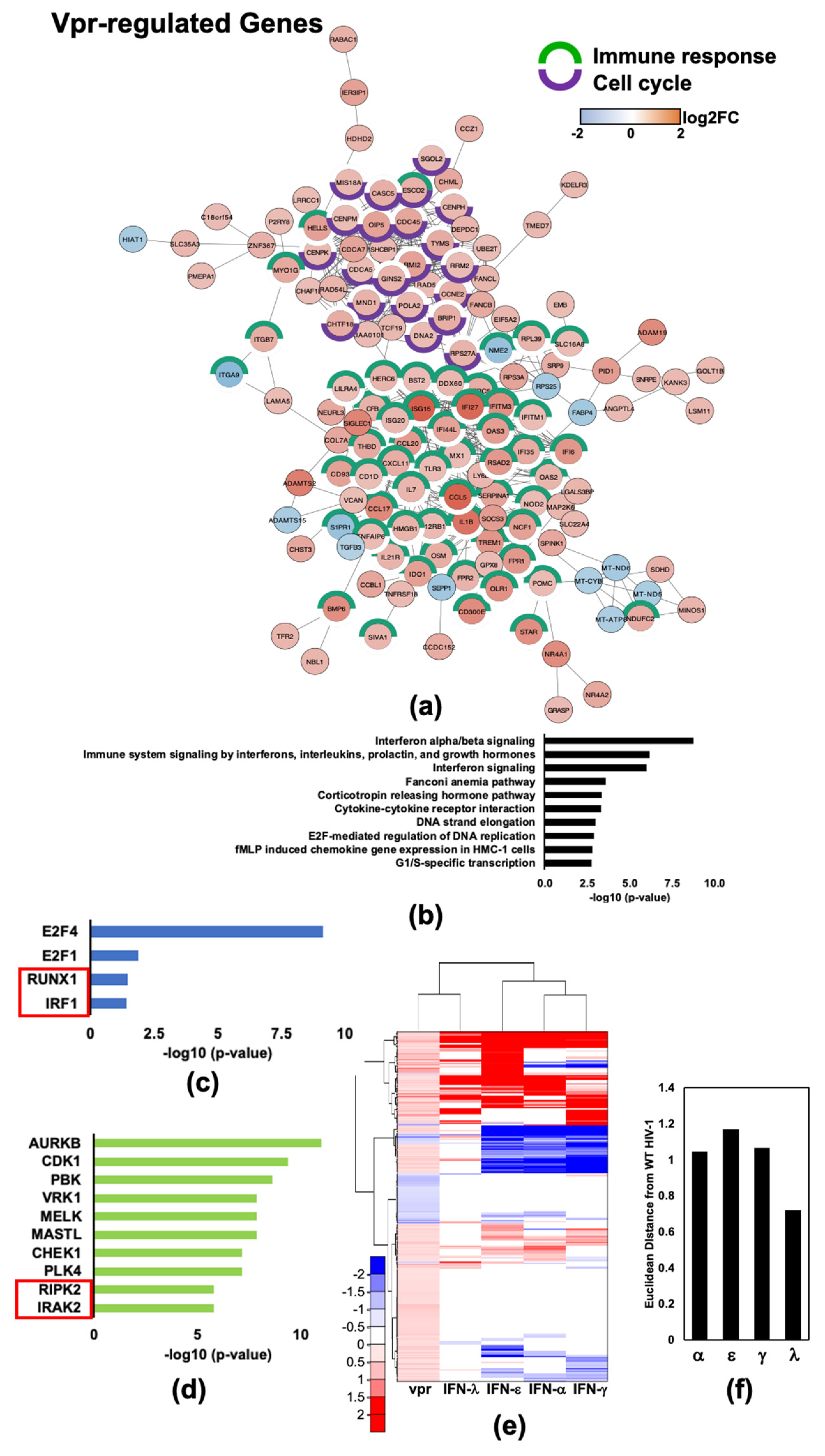
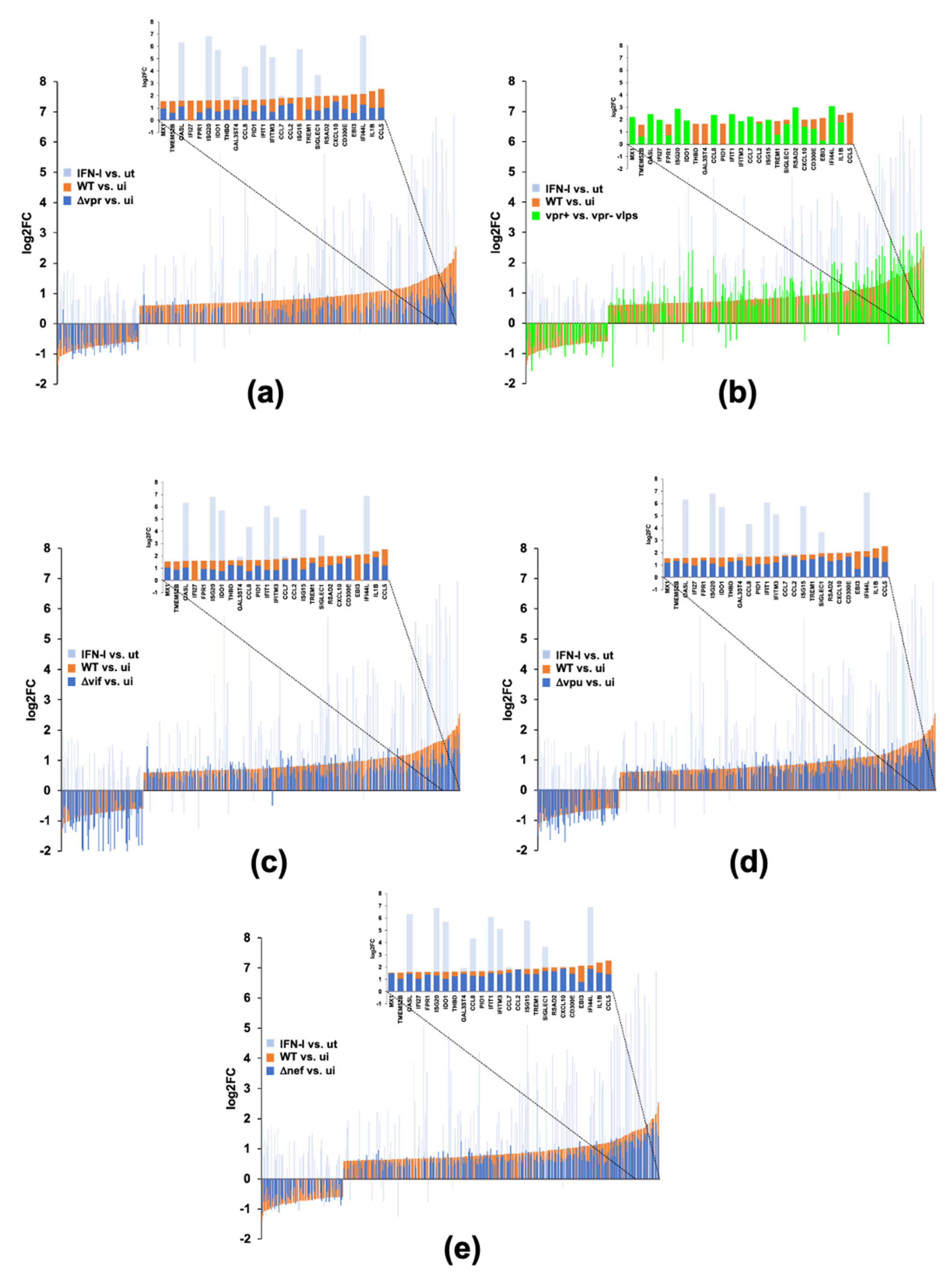
2.5. HIV-1Vpr Induces an Attenuated IFN Response and Regulates Host Cell Cycle Genes in MDMs
2.6. HIV-1 Vpu and Nef Induce Transcriptome Changes Related to Immune Functions
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Altfeld, M.; Gale, M., Jr. Innate immunity against HIV-1 infection. Nat. Immunol. 2015, 16, 554–662. [Google Scholar] [CrossRef] [PubMed]
- Decalf, J.; Desdouits, M.; Rodrigues, V.; Gobert, F.X.; Gentili, M.; Marques-Ladeira, S.; Chamontin, C.; Mougel, M.; Cunha de Alencar, B.; Benaroch, P. Sensing of HIV-1 Entry Triggers a Type I Interferon Response in Human Primary Macrophages. J. Virol. 2017, 91, e00147-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ablasser, A.; Schmid-Burgk, J.L.; Hemmerling, I.; Horvath, G.L.; Schmidt, T.; Latz, E.; Hornung, V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 2013, 503, 530–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broggi, A.; Granucci, F.; Zanoni, I. Type III interferons: Balancing tissue tolerance and resistance to pathogen invasion. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Hou, W.; Wang, X.; Ye, L.; Zhou, L.; Yang, Z.Q.; Riedel, E.; Ho, W.Z. Lambda interferon inhibits human immunodeficiency virus type 1 infection of macrophages. J. Virol. 2009, 83, 3834–3842. [Google Scholar] [CrossRef] [Green Version]
- Stacey, A.R.; Norris, P.J.; Qin, L.; Haygreen, E.A.; Taylor, E.; Heitman, J.; Lebedeva, M.; DeCamp, A.; Li, D.; Grove, D.; et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J. Virol. 2009, 83, 3719–3733. [Google Scholar] [CrossRef] [Green Version]
- Honeycutt, J.B.; Wahl, A.; Baker, C.; Spagnuolo, R.A.; Foster, J.; Zakharova, O.; Wietgrefe, S.; Caro-Vegas, C.; Madden, V.; Sharpe, G.; et al. Macrophages sustain HIV replication in vivo independently of T cells. J. Clin. Investig. 2016, 126, 1353–1366. [Google Scholar] [CrossRef]
- Sattentau, Q.J.; Stevenson, M. Macrophages and HIV-1: An Unhealthy Constellation. Cell Host Microbe 2016, 19, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Szaniawski, M.A.; Spivak, A.M.; Cox, J.E.; Catrow, J.L.; Hanley, T.; Williams, E.; Tremblay, M.J.; Bosque, A.; Planelles, V. SAMHD1 Phosphorylation Coordinates the Anti-HIV-1 Response by Diverse Interferons and Tyrosine Kinase Inhibition. mBio 2018, 9, e00819-18. [Google Scholar] [CrossRef] [Green Version]
- Hardy, G.A.; Sieg, S.; Rodriguez, B.; Anthony, D.; Asaad, R.; Jiang, W.; Mudd, J.; Schacker, T.; Funderburg, N.T.; Pilch-Cooper, H.A.; et al. Interferon-alpha is the primary plasma type-I IFN in HIV-1 infection and correlates with immune activation and disease markers. PLoS ONE 2013, 8, e56527. [Google Scholar] [CrossRef] [Green Version]
- Sandler, N.G.; Bosinger, S.E.; Estes, J.D.; Zhu, R.T.; Tharp, G.K.; Boritz, E.; Levin, D.; Wijeyesinghe, S.; Makamdop, K.N.; del Prete, G.Q.; et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature 2014, 511, 601–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhen, A.; Rezek, V.; Youn, C.; Lam, B.; Chang, N.; Rick, J.; Carrillo, M.; Martin, H.; Kasparian, S.; Syed, P.; et al. Targeting type I interferon-mediated activation restores immune function in chronic HIV infection. J. Clin. Investig. 2016, 127, 260–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Ivashkiv, L.B. IFNgamma: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Strebel, K. HIV accessory proteins versus host restriction factors. Curr. Opin. Virol. 2013, 3, 692–699. [Google Scholar] [CrossRef] [Green Version]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Sheehy, A.M.; Gaddis, N.; Malim, M. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 2003, 9, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Szaniawski, M.A.; Spivak, A.M.; Bosque, A.; Planelles, V. Sex Influences SAMHD1 Activity and Susceptibility to Human Immunodeficiency Virus-1 in Primary Human Macrophages. J. Infect. Dis. 2018, 219, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Harman, A.N.; Lai, J.; Turville, S.; Samarajiwa, S.; Gray, L.; Marsden, V.; Mercier, S.K.; Jones, K.; Nasr, N.; Rustagi, A.; et al. HIV infection of dendritic cells subverts the IFN induction pathway via IRF-1 and inhibits type 1 IFN production. Blood 2011, 118, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schynkel, T.; Szaniawski, M.A.; Spivak, A.M.; Bosque, A.; Planelles, V.; Vandekerckhove, L.; Trypsteen, W. Interferon-Mediated Long Non-Coding RNA Response in Macrophages in the Context of HIV. Int. J. Mol. Sci. 2020, 21, 7741. [Google Scholar] [CrossRef]
- Laoukili, J.; Kooistra, M.R.; Bras, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef]
- Zona, S.; Bella, L.; Burton, M.J.; Nestal de Moraes, G.; Lam, E.W. FOXM1: An emerging master regulator of DNA damage response and genotoxic agent resistance. Biochim. Biophys. Acta 2014, 1839, 1316–1322. [Google Scholar] [CrossRef] [Green Version]
- Clerzius, G.; Gelinas, J.F.; Gatignol, A. Multiple levels of PKR inhibition during HIV-1 replication. Rev. Med. Virol. 2010, 21, 42–53. [Google Scholar] [CrossRef]
- Schmidt, S.V.; Schultze, J.L. New Insights into IDO Biology in Bacterial and Viral Infections. Front. Immunol. 2014, 5, 384. [Google Scholar] [CrossRef] [Green Version]
- Pallotta, M.T.; Rossini, S.; Suvieri, C.; Coletti, A.; Orabona, C.; Macchiarulo, A.; Volpi, C.; Grohmann, U. Indoleamine 2,3-dioxygenase 1 (IDO1): An up-to-date overview of an eclectic immunoregulatory enzyme. FEBS J. 2021, 288, 16086. [Google Scholar] [CrossRef]
- Fernandez, J.; Portilho, D.M.; Danckaert, A.; Munier, S.; Becker, A.; Roux, P.; Zambo, A.; Shorte, S.; Jacob, Y.; Vidalain, P.O.; et al. Microtubule-associated proteins 1 (MAP1) promote human immunodeficiency virus type I (HIV-1) intracytoplasmic routing to the nucleus. J. Biol. Chem. 2015, 290, 4631–4646. [Google Scholar] [CrossRef] [Green Version]
- Malikov, V.; Da Silva, E.S.; Jovasevic, V.; Bennett, G.; Vieira, D.A.D.S.A.; Schulte, B.; Diaz-Griffero, F.; Walsh, D.; Naghavi, M.H. HIV-1 capsids bind and exploit the kinesin-1 adaptor FEZ1 for inward movement to the nucleus. Nat. Commun. 2015, 6, 6660. [Google Scholar] [CrossRef] [PubMed]
- Naghavi, M.H.; Walsh, D. Microtubule Regulation and Function during Virus Infection. J. Virol. 2017, 91, e00538-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshiere, A.; Joly-Beauparlant, C.; Breton, Y.; Ouellet, M.; Raymond, F.; Lodge, R.; Barat, C.; Roy, M.A.; Corbeil, J.; Tremblay, M.J. Global Mapping of the Macrophage-HIV-1 Transcriptome Reveals that Productive Infection Induces Remodeling of Host Cell DNA and Chromatin. Sci. Rep. 2017, 7, 5238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Faust, T.B.; Binning, J.M.; Gross, J.D.; Frankel, A.D. Making Sense of Multifunctional Proteins: Human Immunodeficiency Virus Type 1 Accessory and Regulatory Proteins and Connections to Transcription. Annu. Rev. Virol. 2017, 4, 241–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauter, D.; Kirchhoff, F. Multilayered and versatile inhibition of cellular antiviral factors by HIV and SIV accessory proteins. Cytokine Growth Factor Rev. 2018, 40, 3–12. [Google Scholar] [CrossRef]
- Cingoz, O.; Goff, S.P. HIV-1 Is a Poor Inducer of Innate Immune Responses. mBio 2019, 10, e02834-18. [Google Scholar] [CrossRef] [Green Version]
- Appelberg, K.S.; Wallet, M.A.; Taylor, J.P.; Cash, M.N.; Sleasman, J.W.; Goodenow, M.M. HIV-1 Infection Primes Macrophages Through STAT Signaling to Promote Enhanced Inflammation and Viral Replication. AIDS Res. Hum. Retrovir. 2017, 33, 690–702. [Google Scholar] [CrossRef]
- Johnson, J.S.; Lucas, S.Y.; Amon, L.M.; Skelton, S.; Nazitto, R.; Carbonetti, S.; Sather, D.N.; Littman, D.R.; Aderem, A. Reshaping of the Dendritic Cell Chromatin Landscape and Interferon Pathways during HIV Infection. Cell Host Microbe 2018, 23, 366–381. [Google Scholar] [CrossRef] [Green Version]
- Soper, A.; Kimura, I.; Nagaoka, S.; Konno, Y.; Yamamoto, K.; Koyanagi, Y.; Sato, K. Type I Interferon Responses by HIV-1 Infection: Association with Disease Progression and Control. Front. Immunol. 2018, 8, 1823. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.S.; De Veaux, N.; Rives, A.W.; Lahaye, X.; Lucas, S.Y.; Perot, B.P.; Luka, M.; Garcia-Paredes, V.; Amon, L.M.; Watters, A.; et al. A Comprehensive Map of the Monocyte-Derived Dendritic Cell Transcriptional Network Engaged upon Innate Sensing of HIV. Cell Rep. 2020, 30, 914–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervantes, C.A.; Oliveira, L.M.; Manfrere, K.C.; Lima, J.F.; Pereira, N.Z.; Duarte, A.J.; Sato, M.N. Antiviral factors and type I/III interferon expression associated with regulatory factors in the oral epithelial cells from HIV-1-serodiscordant couples. Sci. Rep. 2016, 6, 25875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, I.T.; Tinago, W.; Sawa, H.; McAndrews, J.; Doak, B.; Prior-Fuller, C.; Sheehan, G.; Lambert, J.S.; Muldoon, E.; Cotter, A.G.; et al. Interferon lambda rs368234815 DeltaG/DeltaG is associated with higher CD4(+):CD8(+) T-cell ratio in treated HIV-1 infection. AIDS Res. Ther. 2020, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Santer, D.M.; Minty, G.E.S.; Golec, D.P.; Lu, J.; May, J.; Namdar, A.; Shah, J.; Elahi, S.; Proud, D.; Joyce, M.; et al. Differential expression of interferon-lambda receptor 1 splice variants determines the magnitude of the antiviral response induced by interferon-lambda 3 in human immune cells. PLoS Pathog. 2020, 16, e1008515. [Google Scholar] [CrossRef]
- Liu, M.Q.; Zhou, D.J.; Wang, X.; Zhou, W.; Ye, L.; Li, J.L.; Wang, Y.Z.; Ho, W.Z. IFN-lambda3 inhibits HIV infection of macrophages through the JAK-STAT pathway. PLoS ONE 2012, 7, e35902. [Google Scholar]
- Read, S.A.; Wijaya, R.; Ramezani-Moghadam, M.; Tay, E.; Schibeci, S.; Liddle, C.; Lam, V.W.T.; Yuen, L.; Douglas, M.W.; Booth, D.; et al. Macrophage Coordination of the Interferon Lambda Immune Response. Front. Immunol. 2019, 10, 2674. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, I.; Porterfield, J.Z.; Gupta, R.K.; Mlcochova, P. Cell Cycle Regulation in Macrophages and Susceptibility to HIV-1. Viruses 2020, 12, 839. [Google Scholar] [CrossRef]
- Mlcochova, P.; Sutherland, K.A.; Watters, S.A.; Bertoli, C.; de Bruin, R.A.; Rehwinkel, J.; Neil, S.J.; Lenzi, G.M.; Kim, B.; Khwaja, A.; et al. A G1-like state allows HIV-1 to bypass SAMHD1 restriction in macrophages. EMBO J. 2017, 36, 604–616. [Google Scholar] [CrossRef]
- Dharan, A.; Campbell, E.M. Role of Microtubules and Microtubule-Associated Proteins in HIV-1 Infection. J. Virol. 2018, 92, e00085-18. [Google Scholar] [CrossRef] [Green Version]
- Hossain, D.; Ferreira Barbosa, J.A.; Cohen, E.A.; Tsang, W.Y. HIV-1 Vpr hijacks EDD-DYRK2-DDB1(DCAF1) to disrupt centrosome homeostasis. J. Biol. Chem. 2018, 293, 9448–9460. [Google Scholar] [CrossRef] [Green Version]
- Coiras, M.; Lopez-Huertas, M.R.; Sanchez del Cojo, M.; Mateos, E.; Alcami, J. Dual role of host cell factors in HIV-1 replication: Restriction and enhancement of the viral cycle. Aids Rev. 2010, 12, 103–112. [Google Scholar] [PubMed]
- Fabryova, H.; Strebel, K. Vpr and Its Cellular Interaction Partners: R We There Yet? Cells 2019, 8, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salamango, D.J.; Harris, R.S. Dual Functionality of HIV-1 Vif in APOBEC3 Counteraction and Cell Cycle Arrest. Front. Microbiol. 2020, 11, 622012. [Google Scholar] [CrossRef]
- Goh, W.C.; Rogel, M.E.; Kinsey, C.M.; Michael, S.F.; Fultz, P.N.; Nowak, M.A.; Hahn, B.H.; Emerman, M. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: A mechanism for selection of Vpr in vivo. Nat. Med. 1998, 4, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Gelbard, H.A.; Roshal, M.; Pursell, S.; Jamieson, B.D.; Planelles, V. Comparison of cell cycle arrest, transactivation, and apoptosis induced by the simian immunodeficiency virus SIVagm and human immunodeficiency virus type 1 vpr genes. J. Virol. 2001, 75, 3791–3801. [Google Scholar] [CrossRef] [Green Version]
- Jager, S.; Kim, D.Y.; Hultquist, J.F.; Shindo, K.; LaRue, R.S.; Kwon, E.; Li, M.; Anderson, B.D.; Yen, L.; Stanley, D.; et al. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 2011, 481, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Kwon, E.; Hartley, P.D.; Crosby, D.C.; Mann, S.; Krogan, N.J.; Gross, J.D. CBFbeta stabilizes HIV Vif to counteract APOBEC3 at the expense of RUNX1 target gene expression. Mol. Cell. 2013, 49, 632–644. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, E.J.D.; Williamson, J.C.; Sienkiewicz, A.; Naamati, A.; Matheson, N.J.; Lehner, P.J. Promiscuous Targeting of Cellular Proteins by Vpr Drives Systems-Level Proteomic Remodeling in HIV-1 Infection. Cell. Rep. 2019, 27, 1579–1596. [Google Scholar] [CrossRef] [Green Version]
- Laguette, N.; Bregnard, C.; Hue, P.; Basbous, J.; Yatim, A.; Larroque, M.; Kirchhoff, F.; Constantinou, A.; Sobhian, B.; Benkirane, M. Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 2014, 156, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Bieniasz, P.D. HIV-1 Vpr induces cell cycle arrest and enhances viral gene expression by depleting CCDC137. Elife 2020, 9, e55806. [Google Scholar] [CrossRef]
- Romani, B.; Shaykh Baygloo, N.; Aghasadeghi, M.R.; Allahbakhshi, E. HIV-1 Vpr Protein Enhances Proteasomal Degradation of MCM10 DNA Replication Factor through the Cul4-DDB1[VprBP] E3 Ubiquitin Ligase to Induce G2/M Cell Cycle Arrest. J. Biol. Chem. 2015, 290, 17380–17389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planelles, V.; Jowett, J.B.; Li, Q.X.; Xie, Y.; Hahn, B.; Chen, I.S. Vpr-induced cell cycle arrest is conserved among primate lentiviruses. J. Virol. 1996, 70, 2516–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, M.C.; Stucky, M.; Wakefield, C.; Melott, J.M.; Akbani, R.; Weinstein, J.N.; Broom, B.M. Interactive Clustered Heat Map Builder: An easy web-based tool for creating sophisticated clustered heat maps. F1000Research 2019, 8, 1750. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, L.J.; Szaniawski, M.A.; Williams, E.S.C.P.; Coiras, M.; Hanley, T.M.; Planelles, V. HIV-1 Accessory Proteins Impart a Modest Interferon Response and Upregulate Cell Cycle-Related Genes in Macrophages. Pathogens 2022, 11, 163. https://doi.org/10.3390/pathogens11020163
Martins LJ, Szaniawski MA, Williams ESCP, Coiras M, Hanley TM, Planelles V. HIV-1 Accessory Proteins Impart a Modest Interferon Response and Upregulate Cell Cycle-Related Genes in Macrophages. Pathogens. 2022; 11(2):163. https://doi.org/10.3390/pathogens11020163
Chicago/Turabian StyleMartins, Laura J., Matthew A. Szaniawski, Elizabeth S. C. P. Williams, Mayte Coiras, Timothy M. Hanley, and Vicente Planelles. 2022. "HIV-1 Accessory Proteins Impart a Modest Interferon Response and Upregulate Cell Cycle-Related Genes in Macrophages" Pathogens 11, no. 2: 163. https://doi.org/10.3390/pathogens11020163