
Dysbiosis in Head and Neck Cancer: Determining Optimal Sampling Site for Oral Microbiome Collection

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Participants
2.3. Sample Collection
2.4. DNA Extraction
2.5. Sequencing, Library Preparation and Analysis
2.6. Statistical Analysis
3. Results
3.1. Study Population and Demographics
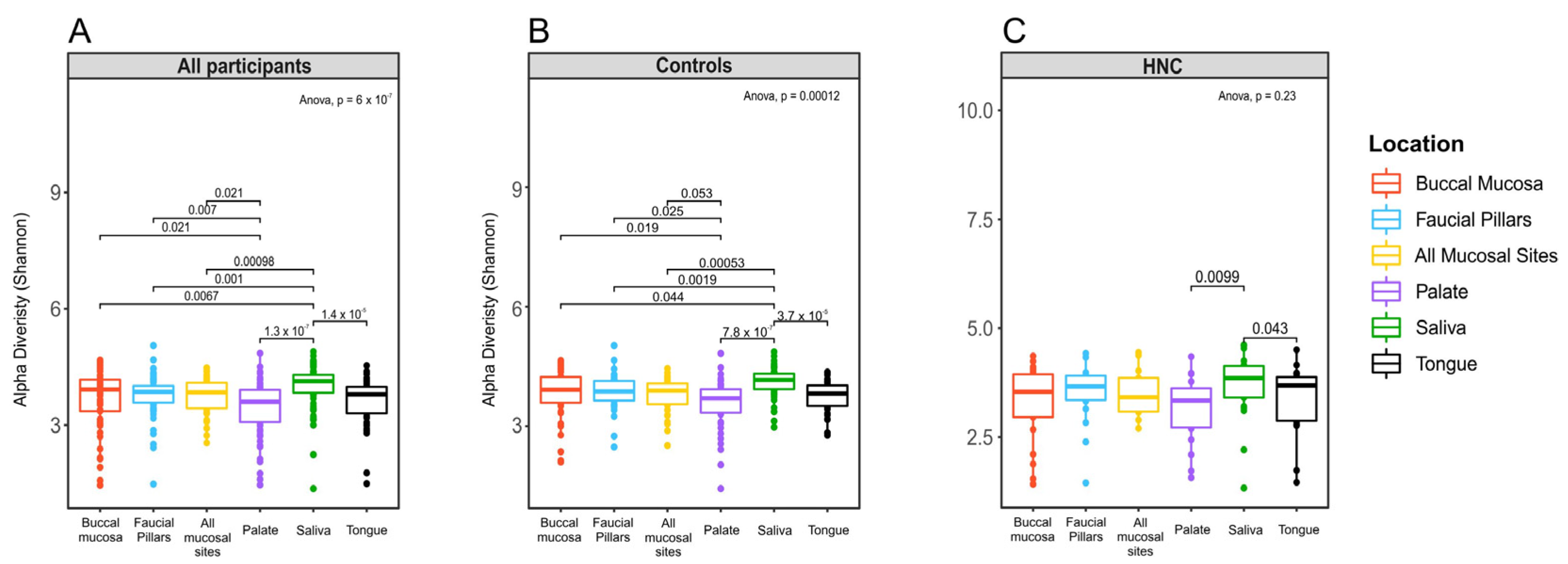
3.2. Comparison of Oral Microbiome Diversity between Distinct Oral sites
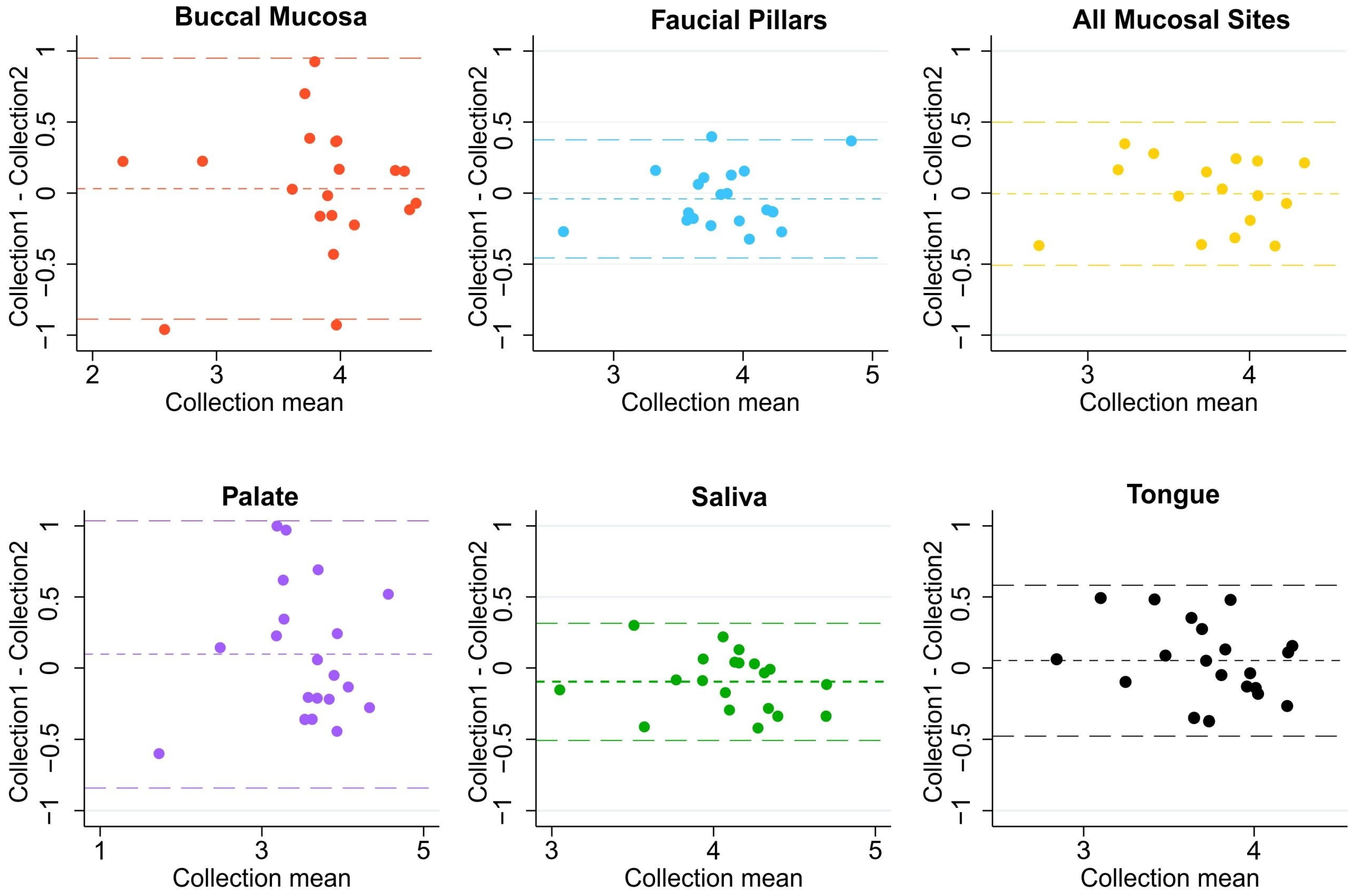
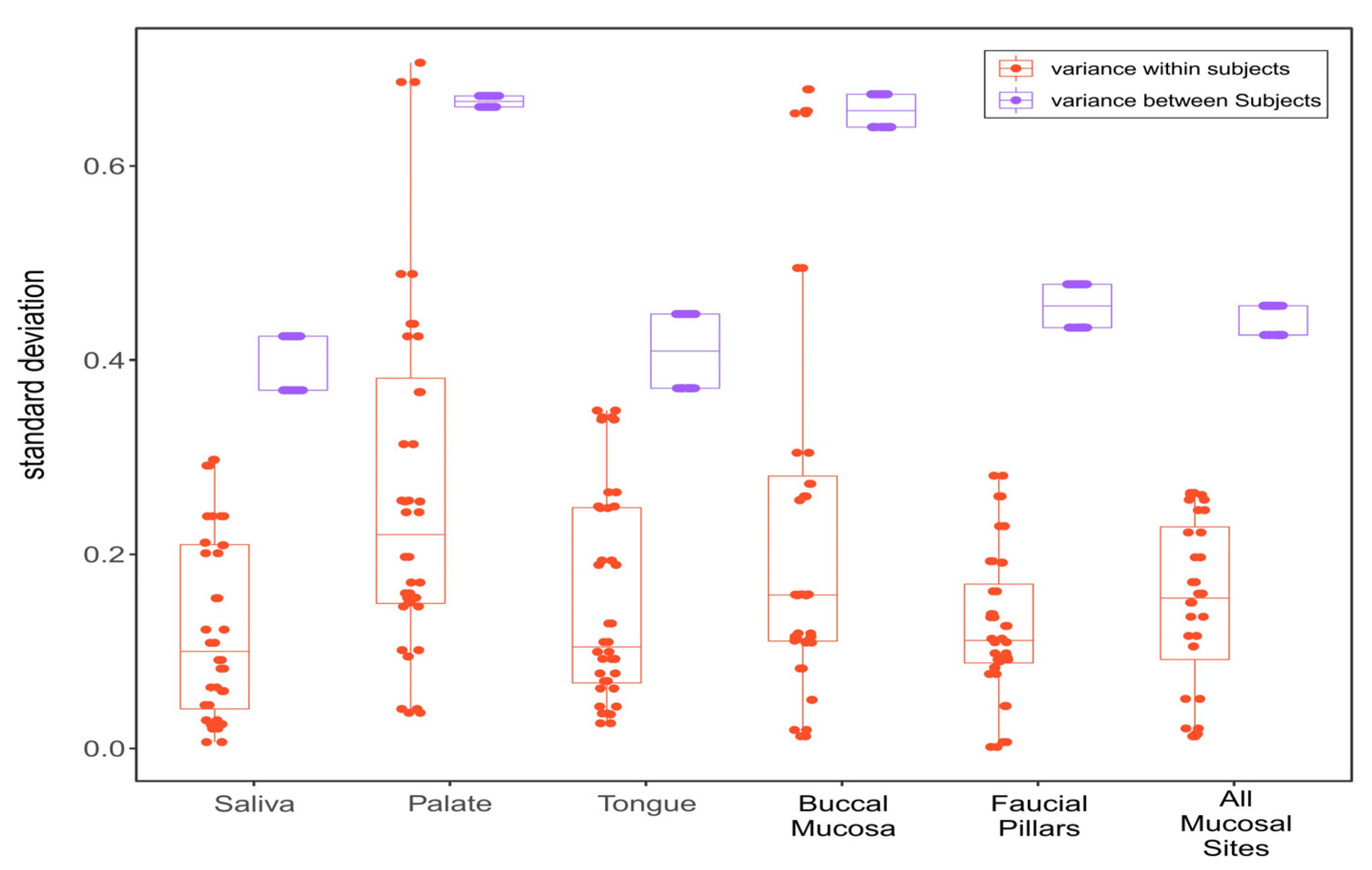
3.3. Within-Subject and between-Subject Variation
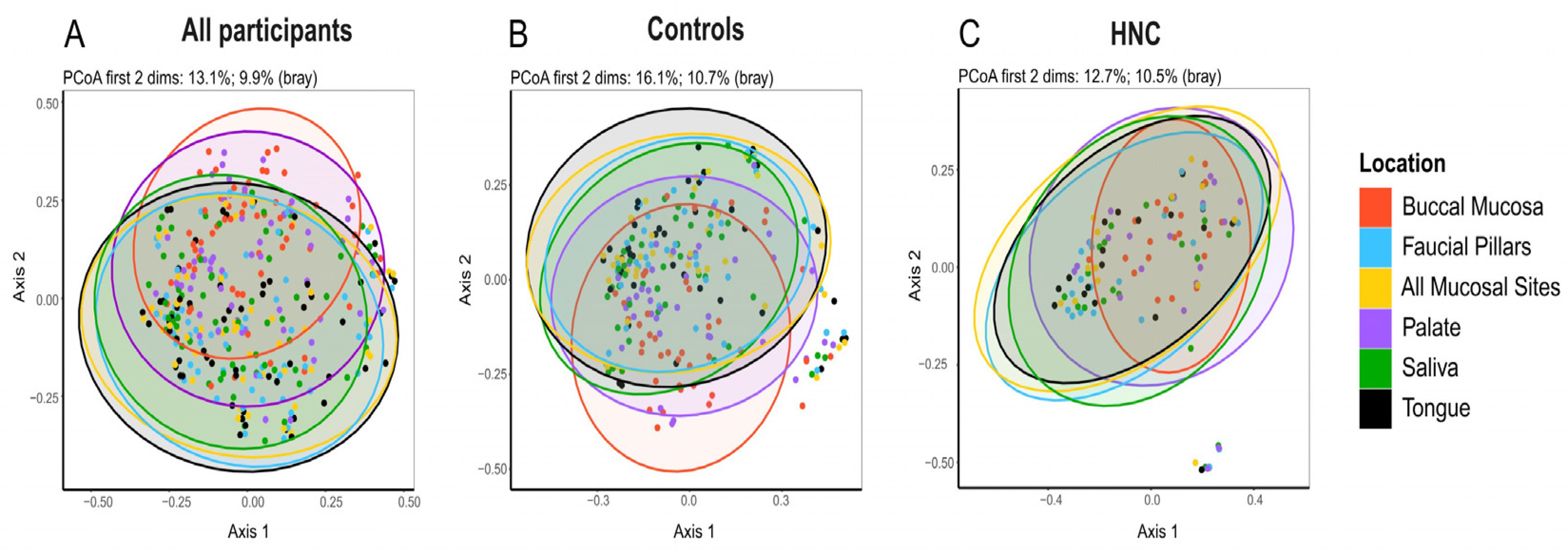
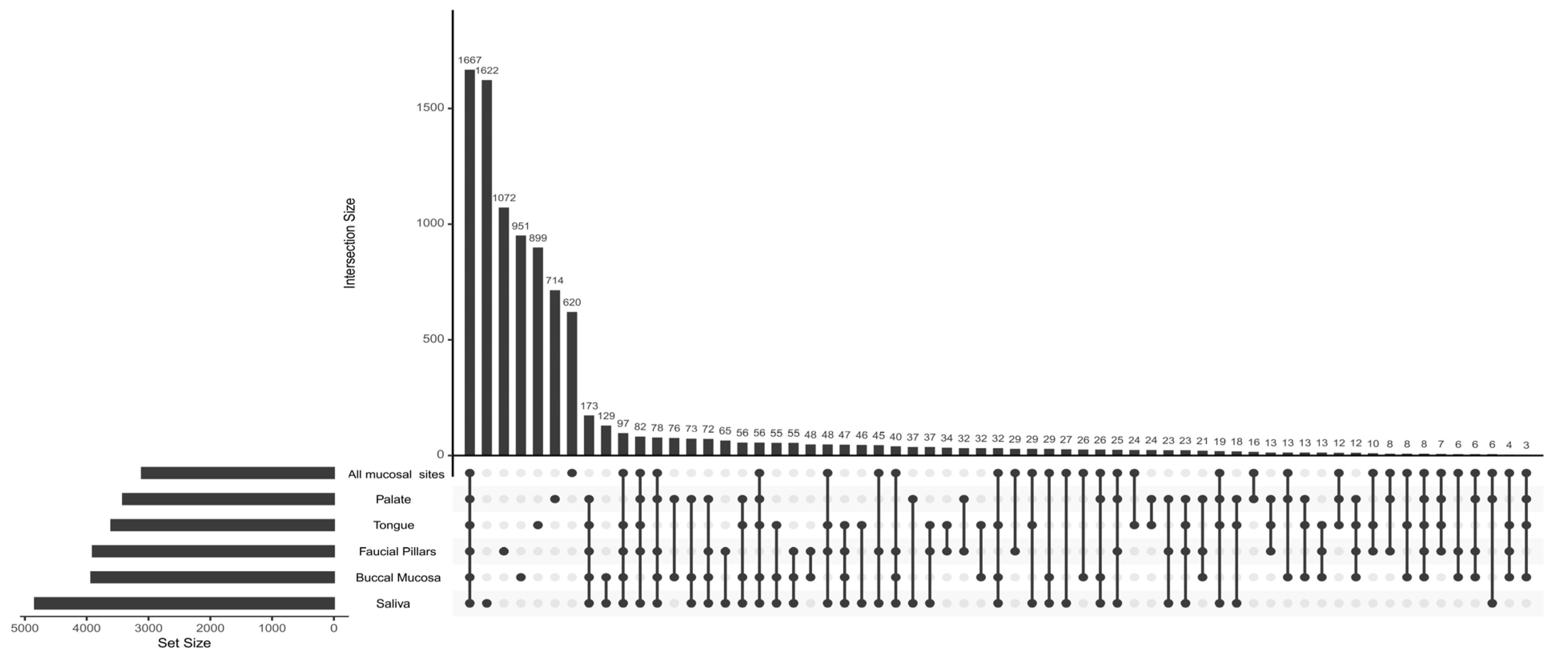
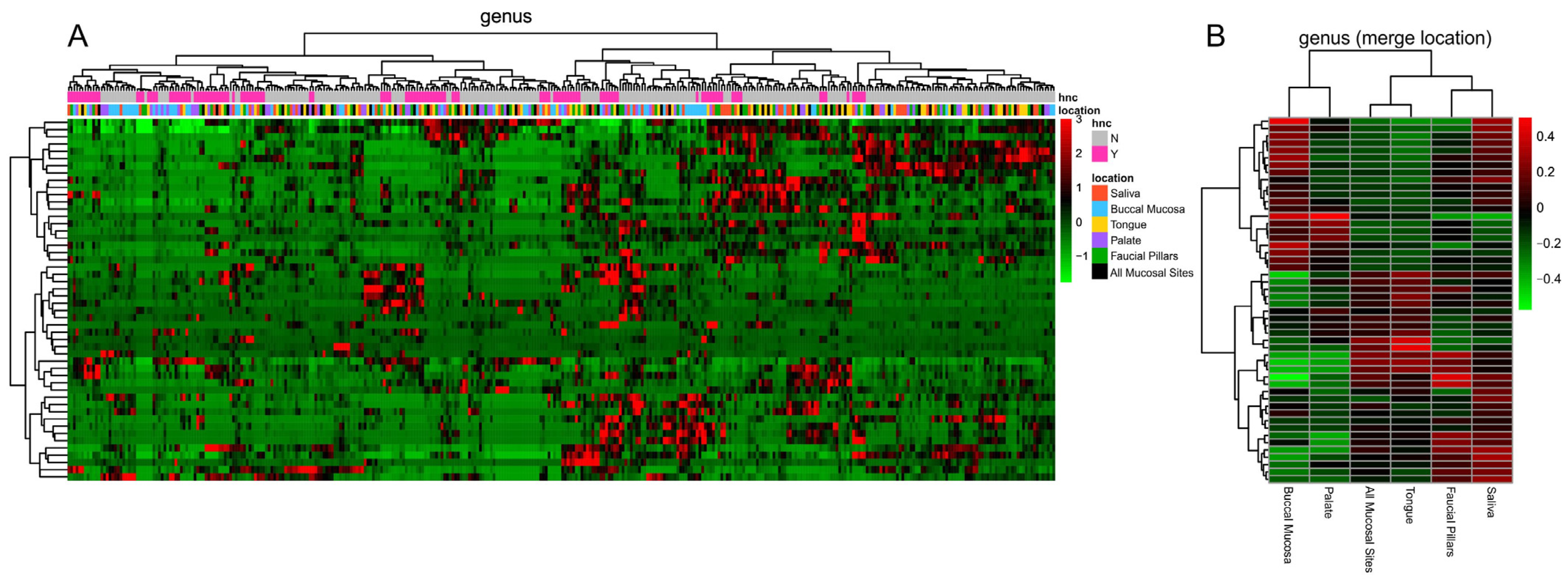
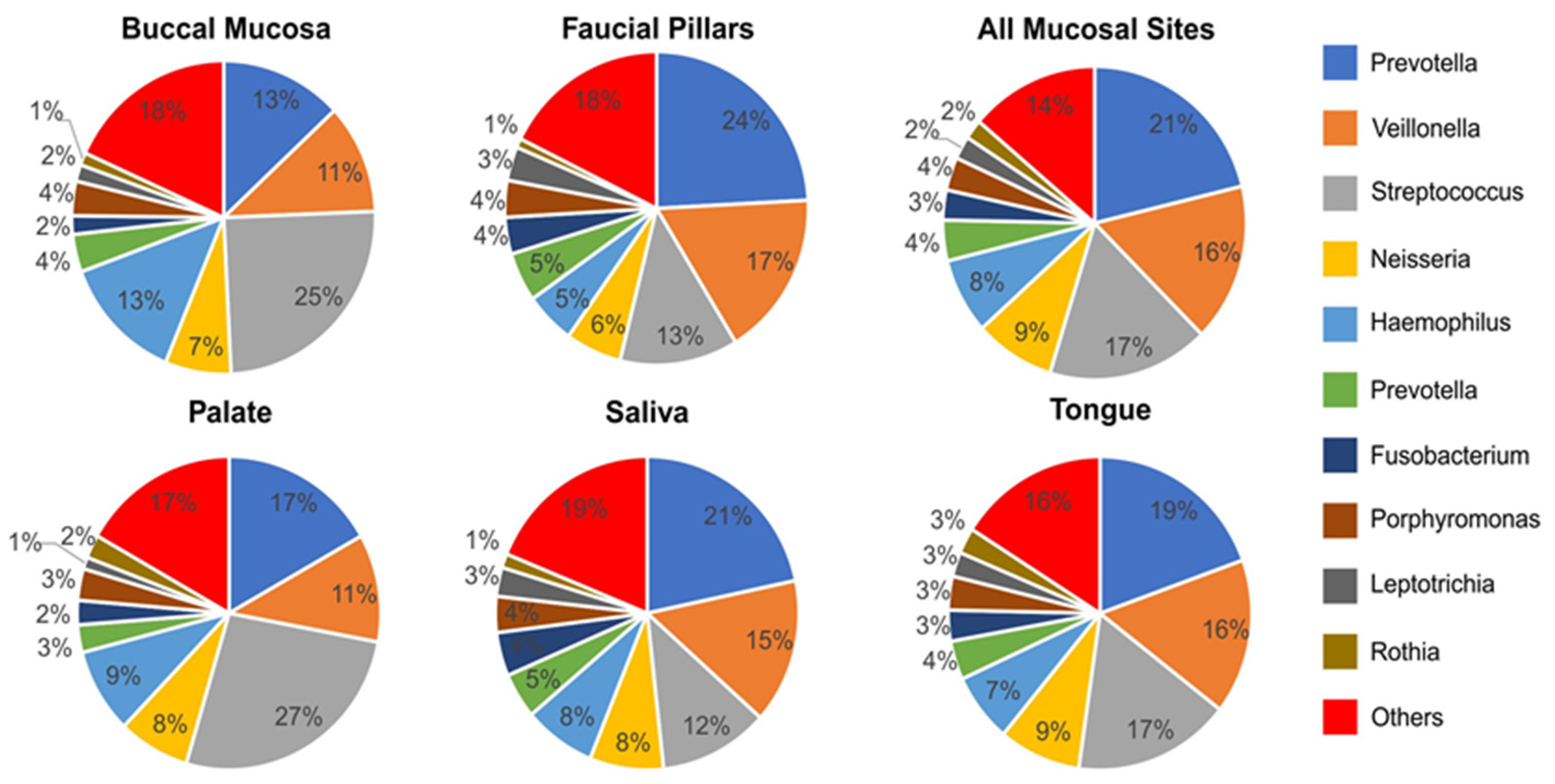
3.4. Oral Microbiome Profile of Saliva and Other Mucosal Sites
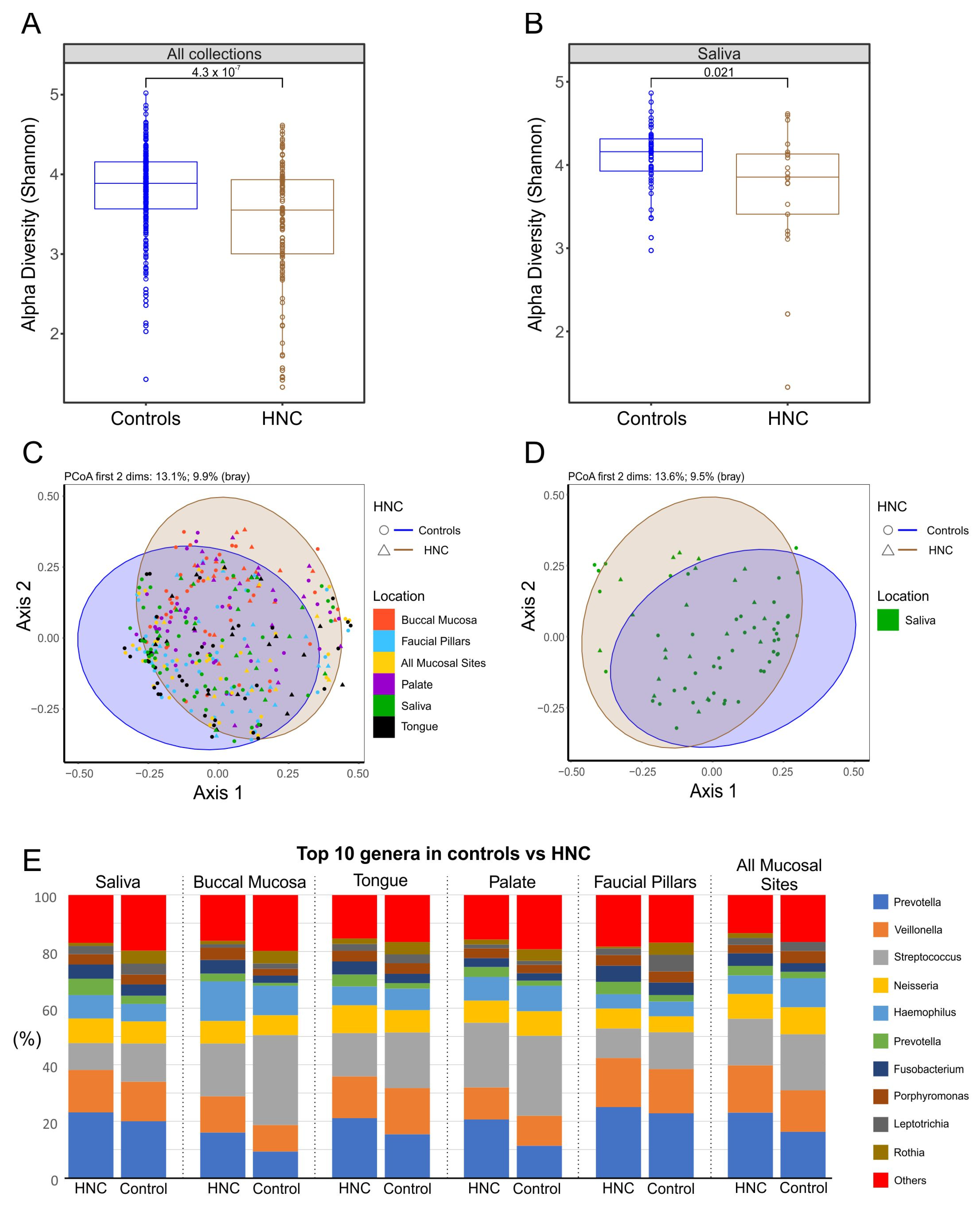
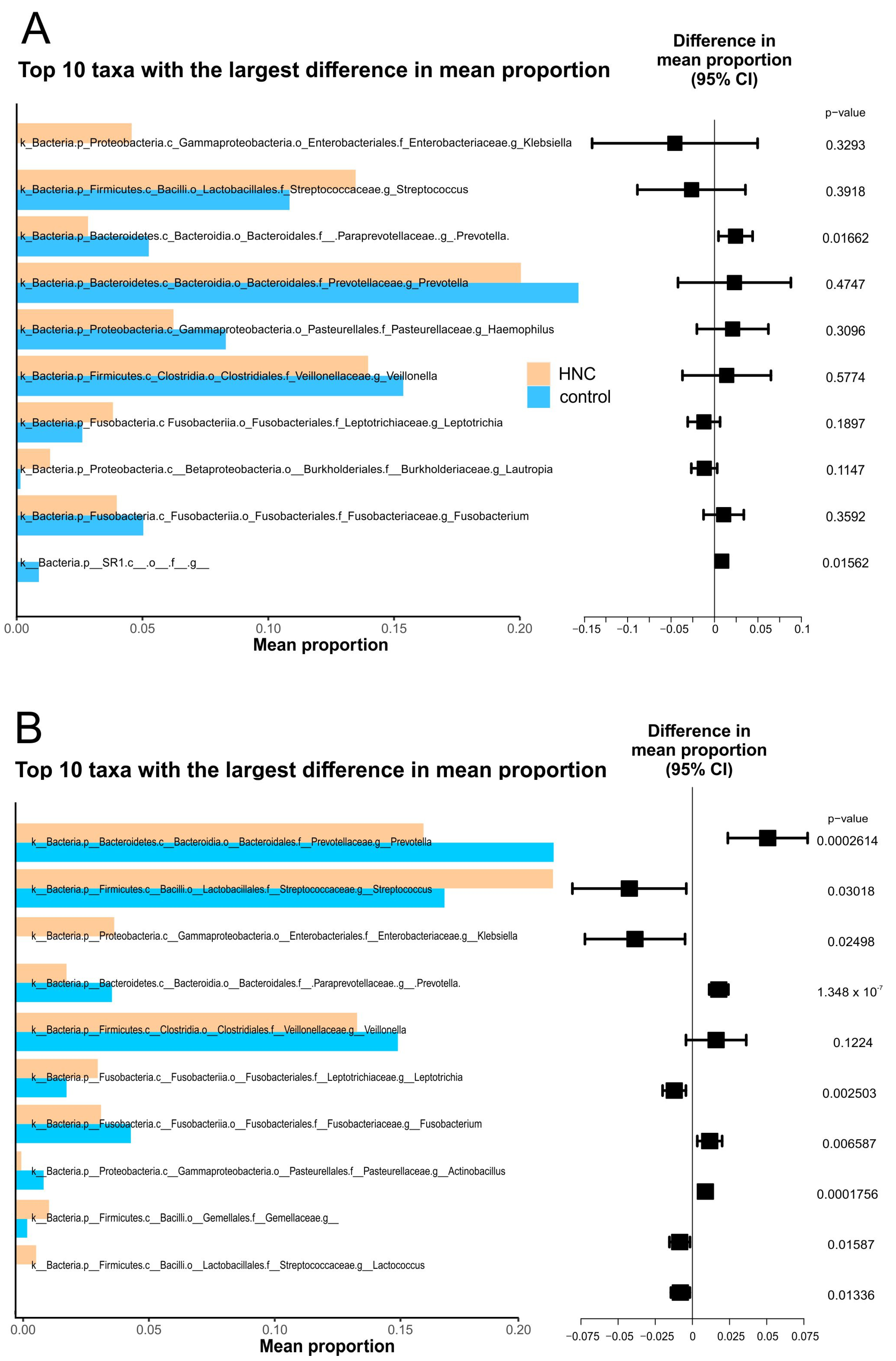
3.5. Comparison of Oral Microbiome between Healthy Controls and HNC
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Warnakulasuriya, S. Global epidemiology of oral and oropharyngeal cancer. Oral. Oncol. 2009, 45, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Australian Institute of Health and Welfare. Cancer in Australia: Actual incidence data from 1982 to 2013 and mortality data from 1982 to 2014 with projections to 2017. Asia Pac. J. Clin. Oncol. 2018, 14, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Alsahafi, E.; Begg, K.; Amelio, I.; Raulf, N.; Lucarelli, P.; Sauter, T.; Tavassoli, M. Clinical update on head and neck cancer: Molecular biology and ongoing challenges. Cell Death Dis. 2019, 10, 540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Tian, T.; Wei, Z.; Peck, K.N.; Shih, N.; Chalian, A.A.; O’Malley, B.W.; Weinstein, G.S.; Feldman, M.D.; Alwine, J.; et al. Microbial Signatures Associated with Oropharyngeal and Oral Squamous Cell Carcinomas. Sci. Rep. 2017, 7, 4036. [Google Scholar] [CrossRef] [Green Version]
- Gong, H.-L.; Shi, Y.; Zhou, L.; Wu, C.-P.; Cao, P.-Y.; Tao, L.; Xu, C.; Hou, D.-S.; Wang, Y.-Z. The Composition of Microbiome in Larynx and the Throat Biodiversity between Laryngeal Squamous Cell Carcinoma Patients and Control Population. PLoS ONE 2013, 8, e66476. [Google Scholar] [CrossRef] [PubMed]
- Mager, D.; Haffajee, A.; Devlin, P.; Norris, C.; Posner, M.R.; Goodson, J. The salivary microbiota as a diagnostic indicator of oral cancer: A descriptive, non-randomized study of cancer-free and oral squamous cell carcinoma subjects. J. Transl. Med. 2005, 3, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, B.; Kuczynski, J.; Bhattacharya, A.; Huey, B.; Corby, P.M.; Queiroz, E.L.S.; Nightingale, K.; Kerr, A.R.; DeLacure, M.D.; Veeramachaneni, R.; et al. Changes in abundance of oral microbiota associated with oral cancer. PLoS ONE 2014, 9, e98741. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Moissl-Eichinger, C.; Perras, A.; Koskinen, K.; Tomazic, P.V.; Thurnher, D. The salivary microbiome as an indicator of carcinogenesis in patients with oropharyngeal squamous cell carcinoma: A pilot study. Sci. Rep. 2017, 7, 5867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.-J.; Shao, Z.-Y.; Wang, Q.; Jiang, Y.-T.; Ma, R.; Tang, Z.-S.; Liu, Z.; Liang, J.-P.; Huang, Z.-W. Exploring the dynamic core microbiome of plaque microbiota during head-and-neck radiotherapy using pyrosequencing. PLoS ONE 2013, 8, e56343. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Teng, F.; Huang, S.; Lin, Z.; Yuan, X.; Zeng, X.; Yang, F. Changes of saliva microbiota in nasopharyngeal carcinoma patients under chemoradiation therapy. Arch. Oral Biol. 2014, 59, 176–186. [Google Scholar] [CrossRef]
- Belazi, M.; Velegraki, A.; Koussidou-Eremondi, T.; Andreadis, D.; Hini, S.; Arsenis, G.; Eliopoulou, C.; Destouni, E.; Oral, D.A. Candida isolates in patients undergoing radiotherapy for head and neck cancer: Prevalence, azole susceptibility profiles and response to antifungal treatment. Oral Microbiol. Immunol. 2004, 19, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Sonalika, W.G.; Tayaar, S.A.; Bhat, K.G.; Patil, B.; Muddapur, M. Oral microbial carriage in oral squamous cell carcinoma patients at the time of diagnosis and during radiotherapy—A comparative study. Oral. Oncol. 2012, 48, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Casariego, A.; Fernandez-Natal, M.I.; Calleja-Fernández, A.; Parras-Padilla, T. Nutritional, Microbiological, and Therapeutic Factors Related to Mucositis in Head and Neck Cancer Patients: A Cohort Study. Nutr. Hosp. 2015, 32, 1208–1213. [Google Scholar] [PubMed]
- Le Bars, P.; Matamoros, S.; Montassier, E.; Le Vacon, F.; Potel, G.; Soueidan, A.; Jordana, F.; De La Cochetière, M.-F. The oral cavity microbiota: Between health, oral disease, and cancers of the aerodigestive tract. Can. J. Microbiol. 2017, 63, 475–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.; Totsika, M.; Morrison, M.; Punyadeera, C. Oral Microbiome: A New Biomarker Reservoir for Oral and Oropharyngeal Cancers. Theranostics 2017, 7, 4313–4321. [Google Scholar] [CrossRef]
- Segata, N.; Haake, S.K.; Mannon, P.; Lemon, K.P.; Waldron, L.; Gevers, D.; Huttenhower, C.; Izard, J. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 2012, 13, R42. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Phillips, S.; Gail, M.H.; Goedert, J.J.; Humphrys, M.; Ravel, J.; Ren, Y.; Caporaso, N.E. Evaluation of Buccal Cell Samples for Studies of Oral Microbiota. Cancer Epidemiol. Biomark. Prev. 2017, 26, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Luo, T.; Srinivasan, U.; Ramadugu, K.; Shedden, K.A.; Neiswanger, K.; Trumble, E.; Li, J.J.; McNeil, D.W.; Crout, R.J.; Weyant, R.J.; et al. Effects of Specimen Collection Methodologies and Storage Conditions on the Short-Term Stability of Oral Microbiome Taxonomy. Appl. Environ. Microbiol. 2016, 82, 5519–5529. [Google Scholar] [CrossRef] [Green Version]
- Vogtmann, E.; Chen, J.; Kibriya, M.G.; Amir, A.; Shi, J.; Chen, Y.; Islam, T.; Eunes, M.; Ahmed, A.; Naher, J.; et al. Comparison of Oral Collection Methods for Studies of Microbiota. Cancer Epidemiol. Biomark. Prev. 2019, 28, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Acharya, A.; Chan, Y.; Kheur, S.; Jin, L.J.; Watt, R.M.; Mattheos, N. Salivary microbiome in non-oral disease: A summary of evidence and commentary. Arch. Oral. Biol. 2017, 83, 169–173. [Google Scholar] [CrossRef]
- Hu, J.; Iragavarapu, S.; Nadkarni, G.N.; Huang, R.; Erazo, M.; Bao, X.; Verghese, D.; Coca, S.; Ahmed, M.K.; Peter, I. Location-Specific Oral Microbiome Possesses Features Associated With CKD. Kidney Int. Rep. 2018, 3, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, K.T.; Thomas, J.G. Oral Microbiome: Contributions to Local and Systemic Infections. Curr. Oral Health Rep. 2016, 3, 45–55. [Google Scholar] [CrossRef]
- Mascitti, M.; Togni, L.; Troiano, G.; Caponio, V.C.A.; Gissi, D.B.; Montebugnoli, L.; Procaccini, M.; Muzio, L.L.; Santarelli, A. Beyond Head and Neck Cancer: The Relationship Between Oral Microbiota and Tumour Development in Distant Organs. Front. Cell Infect. Microbiol. 2019, 9, 232. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, J.M.; Schafer, C.A.; Schafer, J.J.; Farrell, J.J.; Paster, B.J.; Wong, D.T.W. Salivary biomarkers: Toward future clinical and diagnostic utilities. Clin. Microbiol. Rev. 2013, 26, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Costalonga, M.; Herzberg, M.C. The oral microbiome and the immunobiology of periodontal disease and caries. Immunol. Lett. 2014, 162, 22–38. [Google Scholar] [CrossRef] [Green Version]
- Caselli, E.; Fabbri, C.; D’Accolti, M.; Soffritti, I.; Bassi, C.; Mazzacane, S.; Franchi, M. Defining the oral microbiome by whole-genome sequencing and resistome analysis: The complexity of the healthy picture. BMC Microbiol. 2020, 20, 120. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Bisanz, J.E. Importing QIIME2 Artifacts and Associated Data into R Sessions. 2018. Available online: https://github.com/jbisanz/qiime2R (accessed on 14 May 2019).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Mattiello, F.; Verbist, B.; Faust, K.; Raes, J.; Shannon, W.D.; Bijnens, L.; Thas, O. A web application for sample size and power calculation in case-control microbiome studies. Bioinformatics 2016, 32, 2038–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Song, F.; Gu, H.; Wei, X.; Zhang, K.; Zhou, Y.; Luo, H. Comparative Evaluation of the Salivary and Buccal Mucosal Microbiota by 16S rRNA Sequencing for Forensic Investigations. Front. Microbiol. 2022, 13, 777882. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; He, J.; Xue, J.; Wang, Y.; Li, K.; Zhang, K.; Guo, Q.; Liu, X.; Zhou, Y.; Cheng, L.; et al. Oral cavity contains distinct niches with dynamic microbial communities. Environ. Microbiol. 2015, 17, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Esberg, A.; Eriksson, L.; Johansson, I. Site- and Time-Dependent Compositional Shifts in Oral Microbiota Communities. Front. Oral Health 2022, 3, 826996. [Google Scholar] [CrossRef] [PubMed]
- Cabral, D.J.; Wurster, J.I.; Flokas, M.E.; Alevizakos, M.; Zabat, M.; Korry, B.J.; Rowan, A.D.; Sano, W.H.; Andreatos, N.; Ducharme, R.B.; et al. The salivary microbiome is consistent between subjects and resistant to impacts of short-term hospitalization. Sci. Rep. 2017, 7, 11040. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-H.; Chung, S.W.; Auh, Q.-S.; Hong, S.-J.; Lee, Y.-A.; Jung, J.; Lee, G.-J.; Park, H.J.; Shin, S., II; Hong, J.Y. Progress in Oral Microbiome Related to Oral and Systemic Diseases: An Update. Diagnostics 2021, 11, 1283. [Google Scholar] [CrossRef]
- Dong, L.; Yin, J.; Zhao, J.; Ma, S.-R.; Wang, H.-R.; Wang, M.; Chen, W.; Wei, W.-Q. Microbial Similarity and Preference for Specific Sites in Healthy Oral Cavity and Esophagus. Front. Microbiol. 2018, 9, 1603. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Mahurkar, A.; Rahnavard, G.; Crabtree, J.; Orvis, J.; Hall, A.B.; Brady, A.; Creasy, H.H.; McCracken, C.; Giglio, M.G.; et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 2017, 550, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Mark Welch, J.L.; Ramírez-Puebla, S.T.; Borisy, G.G. Oral Microbiome Geography: Micron-Scale Habitat and Niche. Cell Host Microbe 2020, 28, 160–168. [Google Scholar] [CrossRef]
- Baker, J.L.; Hendrickson, E.L.; Tang, X.; Lux, R.; He, X.; Edlund, A.; McLean, J.S.; Shi, W. Klebsiella and Providencia emerge as lone survivors following long-term starvation of oral microbiota. Proc. Natl. Acad. Sci. USA 2019, 116, 8499–8504. [Google Scholar] [CrossRef]
- Simon-Soro, A.; Tomas, I.; Cabrera-Rubio, R.; Catalan, M.; Nyvad, B.; Mira, A. Microbial geography of the oral cavity. J. Dent. Res. 2013, 92, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Eren, A.M.; Borisy, G.G.; Huse, S.M.; Welch, J.L.M. Oligotyping analysis of the human oral microbiome. Proc. Natl. Acad. Sci. USA 2014, 111, E2875–E2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proctor, D.M.; Fukuyama, J.A.; Loomer, P.M.; Armitage, G.C.; Lee, S.A.; Davis, N.M.; Ryder, M.I.; Holmes, S.P.; Relman, D.A. A spatial gradient of bacterial diversity in the human oral cavity shaped by salivary flow. Nat. Commun. 2018, 9, 681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, R.; Nishimoto, Y.; Umezawa, K.; Yama, K.; Aita, Y.; Ichiba, Y.; Murakami, S.; Kakizawa, Y.; Kumagai, T.; Yamada, T.; et al. Comparison of oral microbiome profiles in stimulated and unstimulated saliva, tongue, and mouth-rinsed water. Sci. Rep. 2019, 9, 16124. [Google Scholar] [CrossRef] [Green Version]
- Lenartova, M.; Tesinska, B.; Janatova, T.; Hrebicek, O.; Mysak, J.; Janata, J.; Najmanova, L. The Oral Microbiome in Periodontal Health. Front. Cell Infect. Microbiol. 2021, 11, 629723. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-HNC Controls (n = 27) | Head and Neck Cancer (n = 21) | p | |
|---|---|---|---|
| Age (years) (±SD) | 63.6 (±12.4) | 59.0 (±14.6) | 0.357 |
| Gender(M) | 70.3% | 80.9% | 0.849 |
| Smokers | 22.2% | 38.1% | 0.3788 |
| Drinkers (>12/week) | 38.1% | 38.1% | 0.758 |
| Diabetes | 11.1% | 9.5% | 0.766 |
| Reflux | 3.7% | 4.7% | 0.585 |
| Gastrointestinal | 7.4% | 4.7% | 0.822 |
| Respiratory | 7.4% | - | 0.585 |
| Cancer (previous/Non-HNC) | 3.7% | 19.1% | 0.065 |
| Endocrine | 3.7% | - | 0.884 |
| OSA | - | - | - |
| Location | Controls | HNC | |||
|---|---|---|---|---|---|
| F | Bray p-Value * | F | Bray p-Value * | ||
| Buccal Mucosa | Faucial Pillars | 4.41 | 0.001 | 2.86 | 0.001 |
| All Mucosal Sites | 4.29 | 0.001 | 1.76 | 0.011 | |
| Palate | 1.45 | 0.093 | 0.54 | 0.975 | |
| Saliva | 3.42 | 0.001 | 1.85 | 0.004 | |
| Tongue | 5.35 | 0.001 | 2.62 | 0.001 | |
| Faucial Pillars | All Mucosal Sites | 0.57 | 0.946 | 0.50 | 0.986 |
| Palate | 2.84 | 0.001 | 1.62 | 0.034 | |
| Saliva | 0.76 | 0.755 | 0.31 | 0.999 | |
| Tongue | 0.86 | 0.604 | 0.65 | 0.879 | |
| All mucosal sites | Palate | 2.34 | 0.001 | 0.91 | 0.589 |
| Saliva | 0.93 | 0.488 | 0.46 | 0.997 | |
| Tongue | 0.19 | 1 | 0.47 | 0.988 | |
| Palate | Saliva | 2.82 | 0.001 | 1.15 | 0.256 |
| Tongue | 2.97 | 0.001 | 1.47 | 0.071 | |
| Saliva | Tongue | 1.32 | 0.163 | 0.60 | 0.947 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, D.; Szczesniak, M.; Maclean, J.; Yim, H.C.H.; Zhang, F.; Graham, P.; El-Omar, E.M.; Wu, P. Dysbiosis in Head and Neck Cancer: Determining Optimal Sampling Site for Oral Microbiome Collection. Pathogens 2022, 11, 1550. https://doi.org/10.3390/pathogens11121550
Pandey D, Szczesniak M, Maclean J, Yim HCH, Zhang F, Graham P, El-Omar EM, Wu P. Dysbiosis in Head and Neck Cancer: Determining Optimal Sampling Site for Oral Microbiome Collection. Pathogens. 2022; 11(12):1550. https://doi.org/10.3390/pathogens11121550
Chicago/Turabian StylePandey, Dheeraj, Michal Szczesniak, Julia Maclean, Howard Chi Ho Yim, Fan Zhang, Peter Graham, Emad M. El-Omar, and Peter Wu. 2022. "Dysbiosis in Head and Neck Cancer: Determining Optimal Sampling Site for Oral Microbiome Collection" Pathogens 11, no. 12: 1550. https://doi.org/10.3390/pathogens11121550