

Comparative Genomic Analysis of a Multidrug-Resistant Staphylococcus hominis ShoR14 Clinical Isolate from Terengganu, Malaysia, Led to the Discovery of Novel Mobile Genetic Elements

 ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Phenotypic Antimicrobial Resistance Profile of S. hominis ShoR14
2.2. Genome Properties of Staphylococcus hominis ShoR14
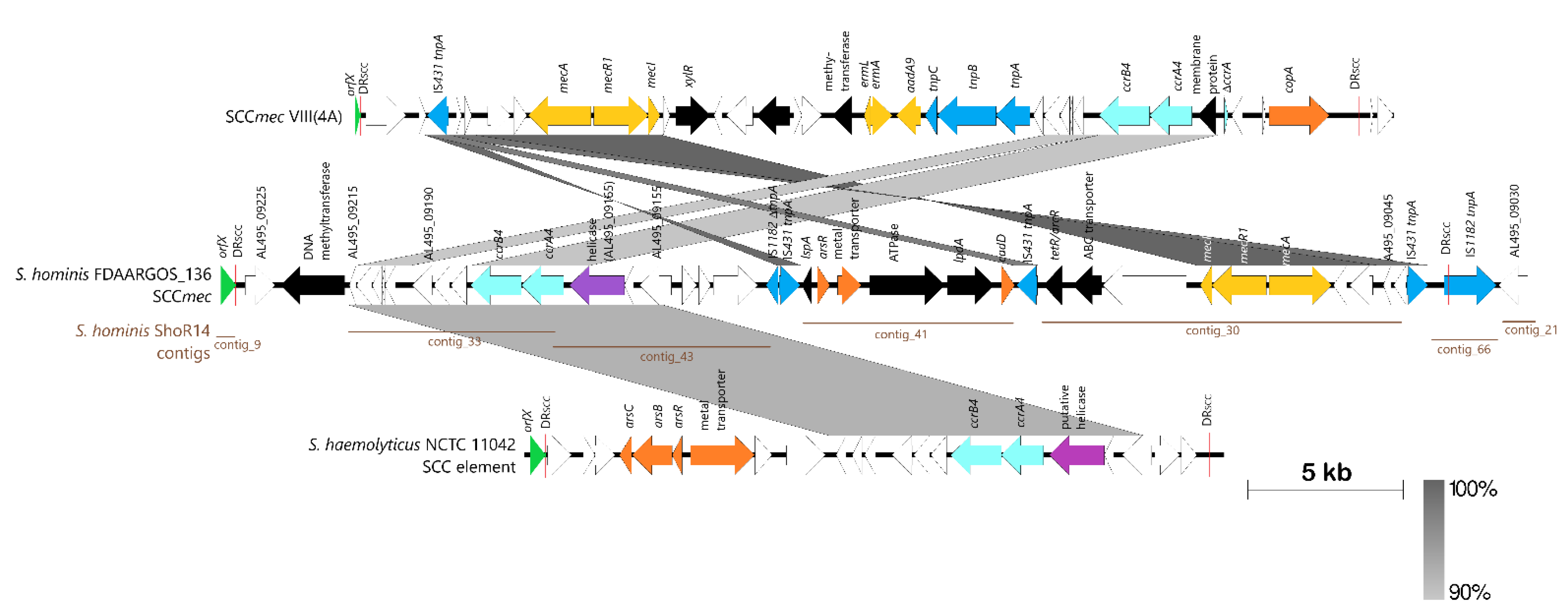
2.3. Prediction of Antimicrobial Resistance and Virulence Genes from the S. hominis ShoR14 Genome Sequence
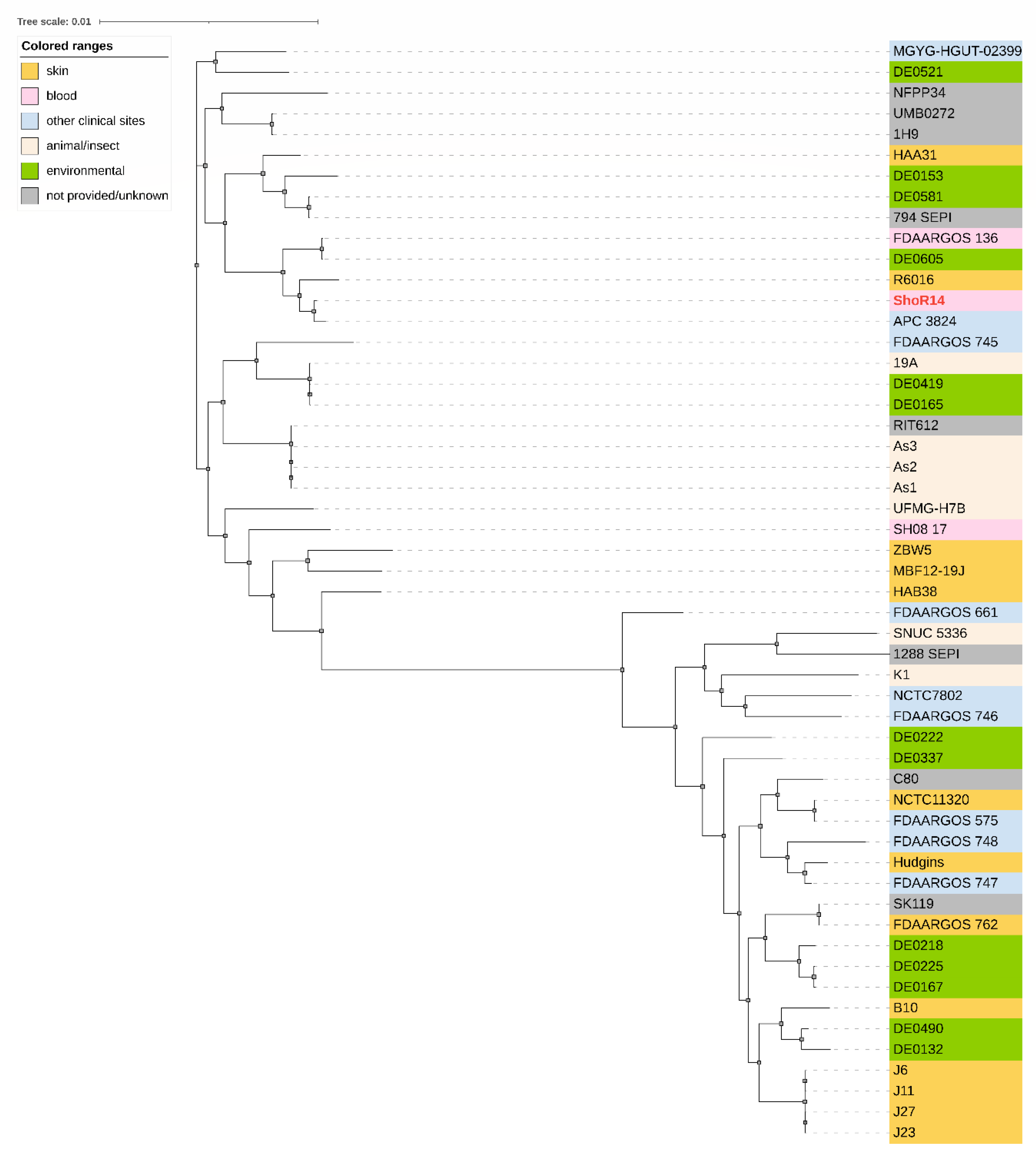
2.4. In Silico Typing and Phylogenetic Analysis of S. hominis ShoR14
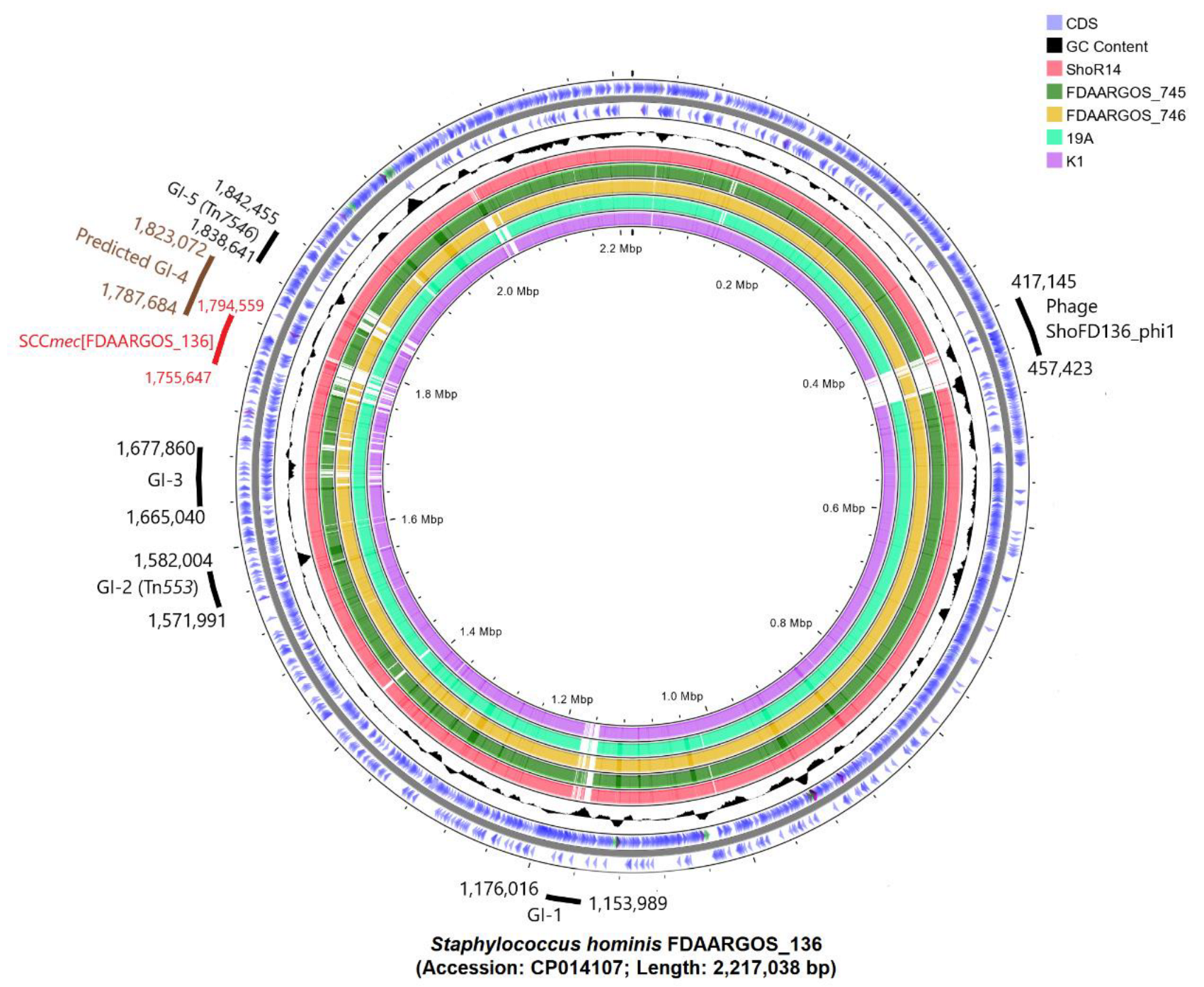
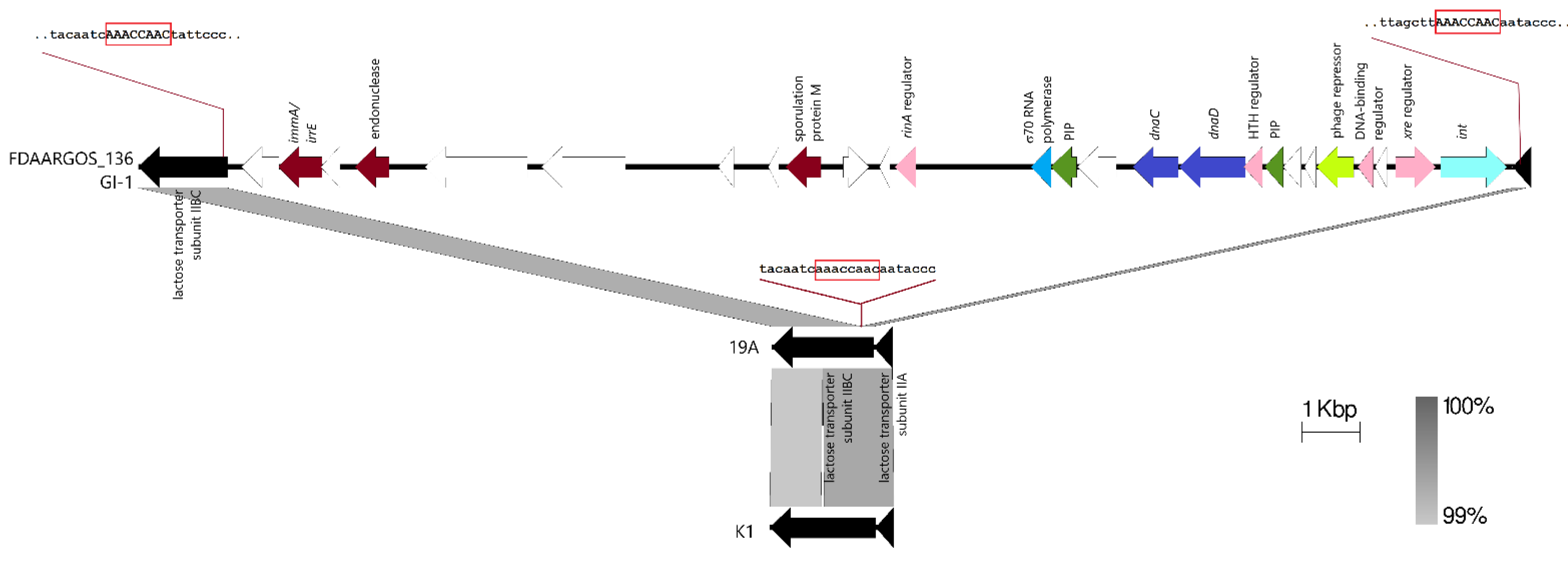
2.5. Comparative Genomic Analysis, Prediction, and Identification of Genomic Islands
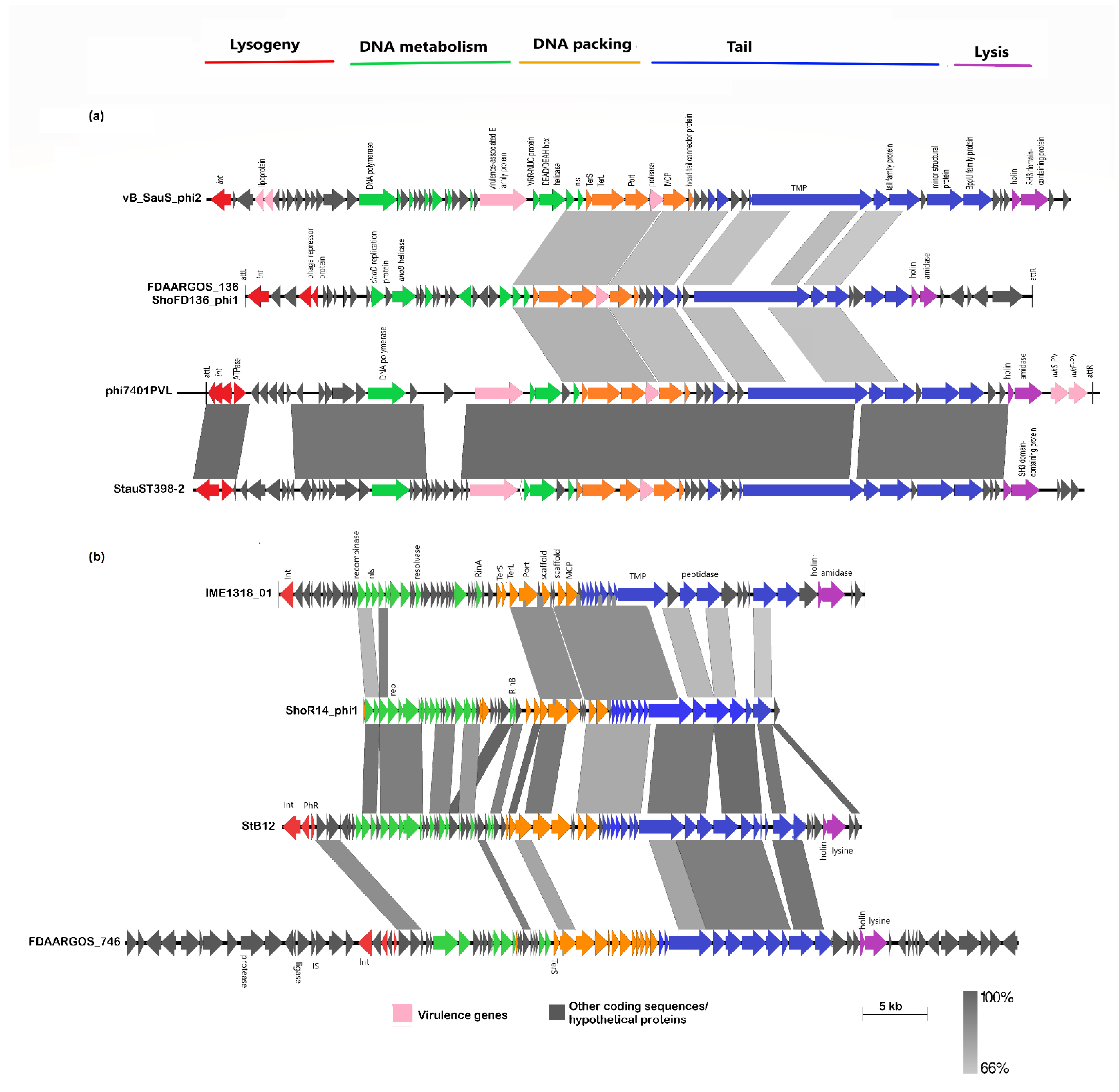
2.6. Identification of S. hominis Prophages
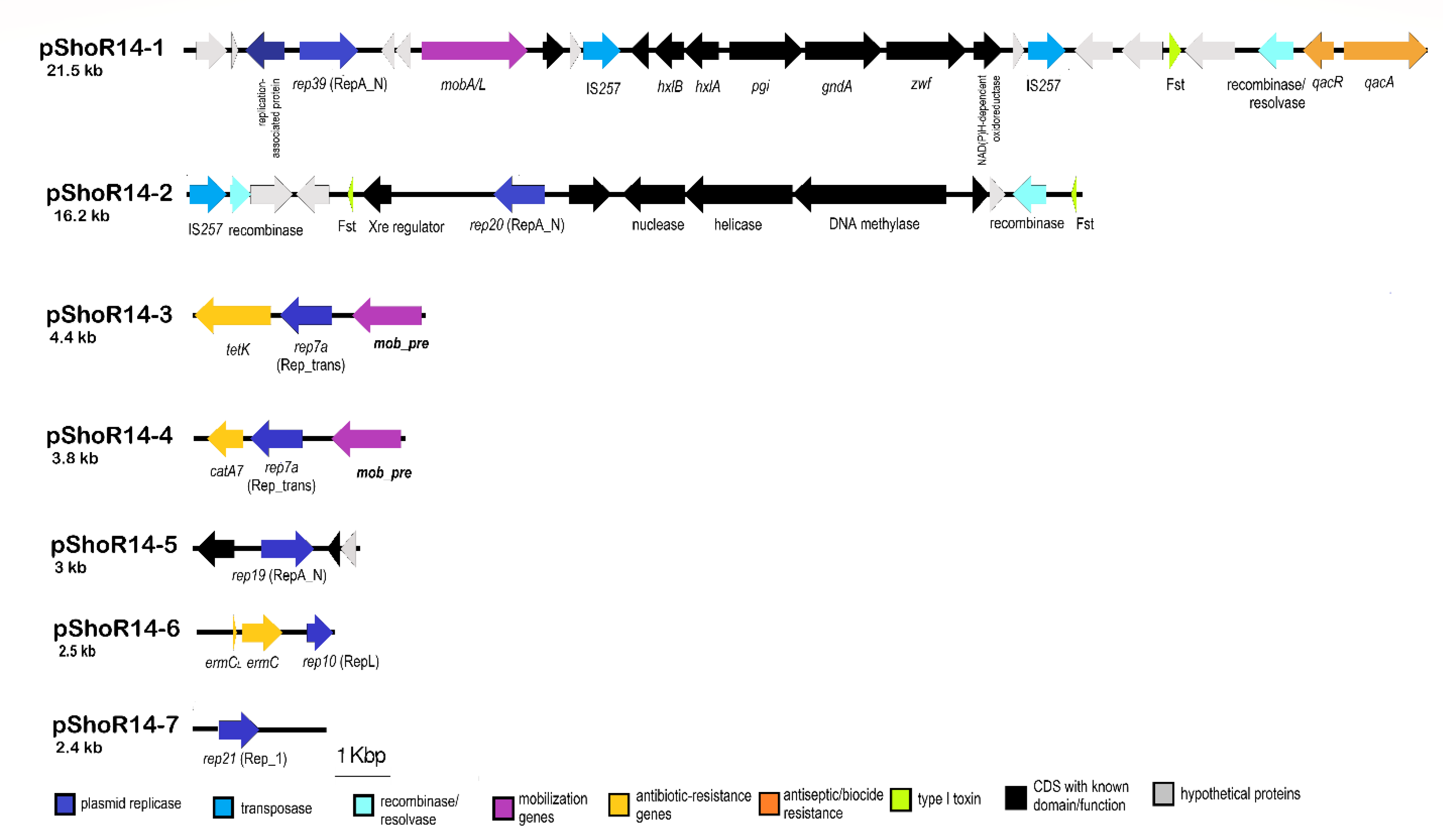
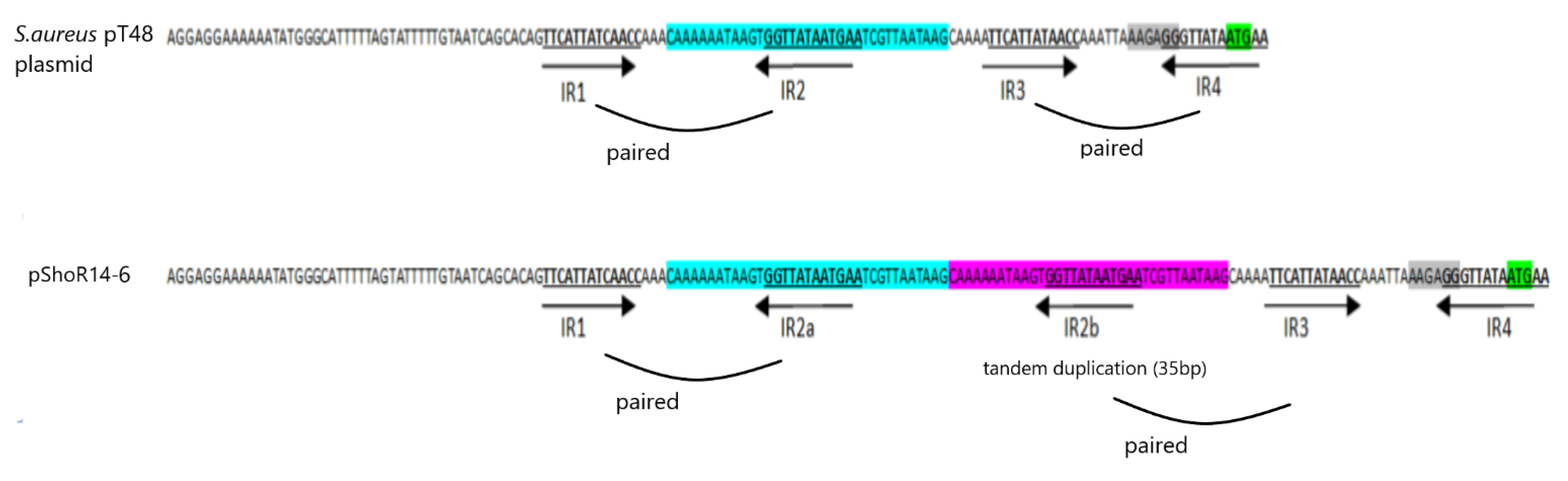
2.7. Identification of Putative Plasmid Sequences in S. hominis ShoR14
3. Conclusions
4. Materials and Methods
4.1. Bacterial Isolate Information
4.2. Antimicrobial Susceptibility and Disc Induction Test
4.3. Whole Genome Sequencing, De Novo Assembly and Annotation
4.4. In Silico Molecular Typing
4.5. Bioinformatics
4.6. Plasmid Identification and Gap Closure
4.7. Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Azevedo, P.A.; Trancesi, R.; Sales, T.; Monteiro, J.; Gales, A.C.; Pignatari, A.C. Outbreak of Staphylococcus hominis Subsp. novobiosepticus Bloodstream Infections in São Paulo City, Brazil. J. Med. Microbiol. 2008, 57, 256–257. [Google Scholar] [CrossRef]
- Becker, K.; Heilmann, C.; Peters, G. Coagulase-Negative Staphylococci. Clin. Microbiol. Rev. 2014, 27, 870–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sychev, Y.V.; Vemulakonda, G.A. Chronic Staphylococcus hominis Endophthalmitis Following Injury with a Retained Intraocular Foreign Body. Eye 2014, 28, 1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza-Olazarán, S.; Morfin-Otero, R.; Rodríguez-Noriega, E.; Llaca-Díaz, J.; Flores-Treviño, S.; González-González, G.M.; Villarreal-Treviño, L.; Garza-González, E. Microbiological and Molecular Characterization of Staphylococcus hominis Isolates from Blood. PLoS ONE 2013, 8, e0061161. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Zheng, B.; Ding, W.; Lv, L.; Ji, J.; Zhang, H.; Xiao, Y.; Li, L. Whole-Genome Sequence of Staphylococcus hominis, an Opportunistic Pathogen. J. Bacteriol. 2012, 194, 4761–4762. [Google Scholar] [CrossRef] [Green Version]
- Bouchami, O.; Ben Hassen, A.; de Lencastre, H.; Miragaia, M. Molecular Epidemiology of Methicillin-Resistant Staphylococcus hominis (MRSHo): Low Clonality and Reservoirs of SCCmec Structural Elements. PLoS ONE 2011, 6, e21940. [Google Scholar] [CrossRef] [PubMed]
- Szemraj, M.; Czekaj, T.; Kalisz, J.; Szewczyk, E.M. Differences in Distribution of MLS Antibiotics Resistance Genes in Clinical Isolates of Staphylococci Belonging to Species: S. epidermidis, S. hominis, S. haemolyticus, S. simulans and S. warneri. BMC Microbiol. 2019, 19, 124. [Google Scholar] [CrossRef] [PubMed]
- Emaneini, M.; Eslampour, M.A.; Sedaghat, H.; Aligholi, M.; Jabalameli, F.; Shahsavan, S.; Taherikalani, M. Characterization of Phenotypic and Genotypic Inducible Macrolide Resistance in Staphylococci in Tehran, Iran. J. Chemother. 2009, 21, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, G.; De Florio, L.; Lorino, G.; Fico, L.; Dicuonzo, G. Macrolide Resistance Genotypes and Phenotypes among Erythromycin-Resistant Clinical Isolates of Staphylococcus aureus and Coagulase-Negative Staphylococci, Italy. FEMS Immunol. Med. Microbiol. 2009, 55, 62–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lina, G.; Quaglia, A.; Reverdy, M.E.; Leclercq, R.; Vandenesch, F.; Etienne, J. Distribution of Genes Encoding Resistance to Macrolides, Lincosamides, and Streptogramins among Staphylococci. Antimicrob. Agents Chemother. 1999, 43, 1062–1066. [Google Scholar] [CrossRef]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-Resistant, Extensively Drug-Resistant and Pandrug-Resistant Bacteria: An International Expert Proposal for Interim Standard Definitions for Acquired Resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding Data and Analysis Capabilities. Nucleic Acids Res. 2020, 48, D606–D612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trzcinski, K.; Cooper, B.S.; Hryniewicz, W.; Dowson, C.G. Expression of Resistance to Tetracyclines in Strains of Methicillin-Resistant Staphylococcus aureus. J. Antimicrob. Chemother. 2000, 45, 763–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guay, G.G.; Khan, S.A.; Rothstein, D.M. The Tet(K) Gene of Plasmid PT181 of Staphylococcus aureus Encodes an Efflux Protein That Contains 14 Transmembrane Helices. Plasmid 1993, 30, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Mojumdar, M.; Khan, S.A. Characterization of the Tetracycline Resistance Gene of Plasmid PT181 of Staphylococcus aureus. J. Bacteriol. 1988, 170, 5522–5528. [Google Scholar] [CrossRef] [Green Version]
- Nesin, M.; Svec, P.; Lupski, J.R.; Godson, G.N.; Kreiswirth, B.; Kornblum, J.; Projan, S.J. Cloning and Nucleotide Sequence of a Chromosomally Encoded Tetracycline Resistance Determinant, TetA(M), from a Pathogenic, Methicillin-Resistant Strain of Staphylococcus Aureus. Antimicrob. Agents Chemother. 1990, 34, 2273–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, S.; Roberts, M.C.; Werckenthin, C.; Pang, Y.; Lange, C. Tetracycline Resistance in Staphylococcus spp. from Domestic Animals. Vet. Microbiol. 1998, 63, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Bismuth, R.; Zilhao, R.; Sakamoto, H.; Guesdon, J.L.; Courvalin, P. Gene Heterogeneity for Tetracycline Resistance in Staphylococcus spp. Antimicrob. Agents Chemother. 1990, 34, 1611–1614. [Google Scholar] [CrossRef] [Green Version]
- Warsa, U.C.; Nonoyama, M.; Ida, T.; Okamoto, R.; Okubo, T.; Shimauchi, C.; Kuga, A.; Inoue, M. Detection of Tet(K) and Tet(M) in Staphylococcus aureus of Asian Countries by the Polymerase Chain Reaction. J. Antibiot. 1996, 49, 1127–1132. [Google Scholar] [CrossRef] [Green Version]
- Hassanzadeh, S.; Ganjloo, S.; Pourmand, M.R.; Mashhadi, R.; Ghazvini, K. Epidemiology of Efflux Pumps Genes Mediating Resistance among Staphylococcus aureus; A Systematic Review. Microb. Pathog. 2020, 139, 103850. [Google Scholar] [CrossRef]
- Gajdács, M. The Continuing Threat of Methicillin-Resistant Staphylococcus aureus. Antibiotics 2019, 8, 52. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.T.; Hung, W.C.; Tsai, J.C.; Leong, K.H.; Chen, H.J.; Hsueh, P.R.; Teng, L.J. Wide Dissemination of SCCfusC in Fusidic Acid-Resistant Coagulase-Negative Staphylococci and Implication for Its Spread to Methicillin-Resistant Staphylococcus aureus in Taiwan. Int. J. Antimicrob. Agents 2018, 51, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Razavi, M.; Marathe, N.P.; Gillings, M.R.; Flach, C.F.; Kristiansson, E.; Joakim Larsson, D.G. Discovery of the Fourth Mobile Sulfonamide Resistance Gene. Microbiome 2017, 5, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, N.; Skurray, R.A. Mobile Elements in the Evolution and Spread of Multiple-Drug Resistance in Staphylococci. Drug Resist. Updat. 1998, 1, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Calkins, S.; Couger, M.B.; Jackson, C.; Zandler, J.; Hudgins, G.C.; Hanafy, R.A.; Budd, C.; French, D.P.; Hoff, W.D.; Youssef, N. Draft Genome Sequence of Staphylococcus hominis Strain Hudgins Isolated from Human Skin Implicates Metabolic Versatility and Several Virulence Determinants. Genomics Data 2016, 10, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.J. Immune Evasion by Staphylococci. Nat. Rev. Microbiol. 2005, 3, 948–958. [Google Scholar] [CrossRef]
- Zhang, L.; Thomas, J.C.; Miragaia, M.; Bouchami, O.; Chaves, F.; D’Azevedo, P.A.; Aanensen, D.M.; de Lencastre, H.; Gray, B.M.; Robinson, D.A. Multilocus Sequence Typing and Further Genetic Characterization of the Enigmatic Pathogen, Staphylococcus hominis. PLoS ONE 2013, 8, e66496. [Google Scholar] [CrossRef] [Green Version]
- Phumthanakorn, N.; Wongsurawat, T.; Jenjaroenpun, P.; Kurilung, A.; Prapasarakul, N. Novel Organization of the Staphylococcal Cassette Chromosome Mec Composite Island in Clinical Staphylococcus haemolyticus and Staphylococcus hominis Subspecies hominis Isolates from Dogs. Microbiol. Spectr. 2022, 10, e0099722. [Google Scholar] [CrossRef]
- Novick, R.P. Pathogenicity Islands and Their Role in Staphylococcal Biology. Gram-Posit. Pathog. 2019, 7, 536–548. [Google Scholar] [CrossRef]
- Krüger, H.; Ji, X.; Wang, Y.; Feßler, A.T.; Wang, Y.; Wu, C.; Schwarz, S. Identification of Tn553, a Novel Tn554-Related Transposon That Carries a Complete BlaZ-BlaR1-BlaI β-Lactamase Operon in Staphylococcus aureus. J. Antimicrob. Chemother. 2021, 76, 2733–2735. [Google Scholar] [CrossRef]
- Zhang, S.; Meyer, R. The Relaxosome Protein MobC Promotes Conjugal Plasmid Mobilization by Extending DNA Strand Separation to the Nick Site at the Origin of Transfer. Mol. Microbiol. 1997, 25, 509–516. [Google Scholar] [CrossRef] [PubMed]
- International Working Group on the Classification of Staphylococcal Cassette Chromosome Elements (IWG-SCC) Classification of Staphylococcal Cassette Chromosome Mec (SCCmec): Guidelines for Reporting Novel SCCmec Elements. Antimicrob. Agents Chemother. 2009, 53, 4961–4967. [CrossRef] [Green Version]
- Uehara, Y. Current Status of Staphylococcal Cassette Chromosome Mec (SCCmec). Antibiotics 2022, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Tansirichaiya, S.; Rahman, M.A.; Roberts, A.P. The Transposon Registry. Mob. DNA 2019, 10, 40. [Google Scholar] [CrossRef]
- Xue, H.; Wu, Z.; Qiao, D.; Tong, C.; Zhao, X. Global Acquisition of Genetic Material from Different Bacteria into the Staphylococcal Cassette Chromosome Elements of a Staphylococcus epidermidis Isolate. Int. J. Antimicrob. Agents 2017, 50, 581–587. [Google Scholar] [CrossRef]
- El Haddad, L.; Roy, J.P.; Khalil, G.E.; St-Gelais, D.; Champagne, C.P.; Labrie, S.; Moineau, S. Efficacy of Two Staphylococcus aureus Phage Cocktails in Cheese Production. Int. J. Food Microbiol. 2016, 217, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Mariem, B.J.J.; Ito, T.; Zhang, M.; Jin, J.; Li, S.; Ilhem, B.B.B.; Adnan, H.; Han, X.; Hiramatsu, K. Molecular Characterization of Methicillin-Resistant Panton-Valentine Leukocidin Positive Staphylococcus aureus Clones Disseminating in Tunisian Hospitals and in the Community. BMC Microbiol. 2013, 13, 2. [Google Scholar] [CrossRef] [Green Version]
- Deghorain, M.; Bobay, L.M.; Smeesters, P.R.; Bousbata, S.; Vermeersch, M.; Perez-Morga, D.; Drèze, P.A.; Rocha, E.P.C.; Touchon, M.; Van Melderen, L. Characterization of Novel Phages Isolated in Coagulase-Negative Staphylococci Reveals Evolutionary Relationships with Staphylococcus aureus Phages. J. Bacteriol. 2012, 194, 5829–5839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deghorain, M.; Van Melderen, L. The Staphylococci Phages Family: An Overview. Viruses 2012, 4, 3316–3335. [Google Scholar] [CrossRef] [Green Version]
- Bosi, E.; Donati, B.; Galardini, M.; Brunetti, S.; Sagot, M.F.; Lió, P.; Crescenzi, P.; Fani, R.; Fondi, M. MeDuSa: A Multi-Draft Based Scaffolder. Bioinformatics 2015, 31, 2443–2451. [Google Scholar] [CrossRef]
- Tian, F.; Li, J.; Li, F.; Tong, Y. Characteristics and Genome Analysis of a Novel Bacteriophage IME1323_01, the First Temperate Bacteriophage Induced from Staphylococcus caprae. Virus Res. 2021, 305, 198569. [Google Scholar] [CrossRef] [PubMed]
- Francia, M.V.; Varsaki, A.; Garcillán-Barcia, M.P.; Latorre, A.; Drainas, C.; De La Cruz, F. A Classification Scheme for Mobilization Regions of Bacterial Plasmids. FEMS Microbiol. Rev. 2004, 28, 79–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, A.J.; Lindsay, J.A. The Distribution of Plasmids That Carry Virulence and Resistance Genes in Staphylococcus aureus Is Lineage Associated. BMC Microbiol. 2012, 12, 104. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.A.; Novick, R.P. Complete Nucleotide Sequence of PT181, a Tetracycline-Resistance Plasmid from Staphylococcus aureus. Plasmid 1983, 10, 251–259. [Google Scholar] [CrossRef]
- Schwarz, F.V.; Perreten, V.; Teuber, M. Sequence of the 50-Kb Conjugative Multiresistance Plasmid PRE25 from Enterococcus faecalis RE25. Plasmid 2001, 46, 170–187. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.; Shen, J.; Wendlandt, S.; Feßler, A.T.; Wang, Y.; Kadlec, K.; Wu, C.-M. Plasmid-Mediated Antimicrobial Resistance in Staphylococci and Other Firmicutes. Microbiol. Spectr. 2014, 2, 421–444. [Google Scholar] [CrossRef] [Green Version]
- Feßler, A.T.; Wang, Y.; Wu, C.; Schwarz, S. Mobile Macrolide Resistance Genes in Staphylococci. Plasmid 2018, 99, 2–10. [Google Scholar] [CrossRef]
- Kuntová, L.; Pantůček, R.; Rájová, J.; Růžičková, V.; Petráš, P.; Mašlaňová, I.; Doškař, J. Characteristics and Distribution of Plasmids in a Clonally Diverse Set of Methicillin-Resistant Staphylococcus aureus Strains. Arch. Microbiol. 2012, 194, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Catchpole, I.; Thomas, C.; Davies, A.; Dyke, K.G.H. The Nucleotide Sequence of Staphylococcus aureus Plasmid PT48 Conferring Inducible Macrolide-Lincosamide-Streptogramin B Resistance and Comparison with Similar Plasmids Expressing Constitutive Resistance. J. Gen. Microbiol. 1988, 134, 697–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lüthje, P.; Schwarz, S. Molecular Analysis of Constitutively Expressed Erm(C) Genes Selected in vitro in the Presence of the Non-Inducers Pirlimycin, Spiramycin and Tylosin. J. Antimicrob. Chemother. 2007, 59, 97–101. [Google Scholar] [CrossRef]
- Gatermann, S.G.; Koschinski, T.; Friedrich, S. Distribution and Expression of Macrolide Resistance Genes in Coagulase-Negative Staphylococci. Clin. Microbiol. Infect. 2007, 13, 777–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczuka, E.; Makowska, N.; Bosacka, K.; Słotwińska, A.; Kaznowski, A. Molecular Basis of Resistance to Macrolides, Lincosamides and Streptogramins in Staphylococcus hominis Strains Isolated from Clinical Specimens. Folia Microbiol. 2016, 61, 143–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.A.; Thomas, J.; Grossman, A.D. The Bacillus Subtilis Conjugative Transposon ICEBs1 Mobilizes Plasmids Lacking Dedicated Mobilization Functions. J. Bacteriol. 2012, 194, 3165–3172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, K.E.; Kwong, S.M.; Firth, N.; Francia, M.V. The RepA _ N Replicons of Gram-Positive Bacteria: A Family of Broadly Distributed but Narrow Host Range Plasmids. Plasmid 2009, 61, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Firth, N.; Apisiridej, S.; Berg, T.; O’Rourke, B.A.; Curnock, S.; Dyke, K.G.H.; Skurray, R.A. Replication of Staphylococcal Multiresistance Plasmids. J. Bacteriol. 2000, 182, 2170–2178. [Google Scholar] [CrossRef] [Green Version]
- Che Hamzah, A.M.; Yeo, C.C.; Puah, S.M.; Chua, K.H.; Rahman, N.I.A.; Abdullah, F.H.; Othman, N.; Chew, C.H. Tigecycline and Inducible Clindamycin Resistance in Clinical Isolates of Methicillin-Resistant Staphylococcus aureus from Terengganu, Malaysia. J. Med. Microbiol. 2019, 68, 1299–1305. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing, 30th ed.; CLSI Supplement M100; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2020. [Google Scholar] [CrossRef]
- The European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters. Version 10.0. 2020. Available online: http://www.eucast.org (accessed on 1 July 2022).
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A Modular and Extensible Implementation of the RAST Algorithm for Building Custom Annotation Pipelines and Annotating Batches of Genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-Access Bacterial Population Genomics: BIGSdb Software, the PubMLST.Org Website and Their Applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and Model-Centric Curation of the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A Comparative Pathogenomic Platform with an Interactive Web Interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Chen, L.; Xiong, Z.; Sun, L.; Yang, J.; Jin, Q. VFDB 2012 Update: Toward the Genetic Diversity and Molecular Evolution of Bacterial Virulence Factors. Nucleic Acids Res. 2012, 40, 641–645. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded Prediction of Genomic Islands for Larger-Scale Datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A Genome Comparison Visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [Green Version]
- Stothard, P.; Grant, J.R.; Van Domselaar, G. Visualizing and Comparing Circular Genomes Using the CGView Family of Tools. Brief. Bioinform. 2019, 20, 1576–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive Visualization of de novo Genome Assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [Green Version]
- Carattoli, A.; Zankari, E.; Garciá-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids Using Plasmidfinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Fasttree: Computing Large Minimum Evolution Trees with Profiles Instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e0009490. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial Class | Resistance Phenotype | Resistance Gene | Mechanism of Resistance | Location of the Resistance Gene |
|---|---|---|---|---|
| β-lactams | Penicillin | blaZ | Antibiotic inactivation enzyme | Chromosomal |
| Cefoxitin, oxacillin | mecA, mecR1, mecI | Antibiotic target alteration | Chromosomal | |
| Fluoroquinolones | Ciprofloxacin, moxifloxacin | norA | Efflux pump conferring antibiotic resistance | Chromosomal |
| Macrolides | Erythromycin | ermC | Antibiotic target alteration | Plasmid |
| Lincosamides | Clindamycin | |||
| Aminoglycosides | Gentamicin | aac(6′)-aph(2″); ant(4′)-Ib; aadD | Antibiotic inactivation enzyme | Chromosomal |
| Folate inhibitors | Co-trimoxazole | sul4, dfrC | Antibiotic target replacement | Chromosomal |
| Fusidanes | Fusidic acid | fusC | Antibiotic target alteration | Chromosomal (SCC element) |
| Tetracyclines | Tetracycline, doxycycline | tetK | Efflux pump conferring antibiotic resistance | Plasmid |
| Phenicols | Chloramphenicol | catA7 | Antibiotic inactivation enzyme | Plasmid |
| Monoxycarbolic acids | Mupirocin | mupA | Antibiotic target alteration | Chromosomal |
| Virulence Factor Classes | Gene | Product |
|---|---|---|
| Adherence | atl | Autolysin |
| ebp | Elastin binding protein | |
| Exoenzyme | lip | Lipase |
| nuc | Thermonuclease | |
| Immune modulation/evasion | orf01763, orf02129, orf02130, orf02131, orf02132, orf02135, orf02139, orf02141 | Capsule biosynthesis proteins |
| capB, capC | Polyglutamic acid capsule |
| Bacterial Species | Position of Tn7546 | Size of Tn7546 (bp) | Length of Target Site Duplication (bp) | Target Site Duplication Sequence | Accession Number |
|---|---|---|---|---|---|
| Staphylococcus hominis ShoR14 | Contig_9: 23,335..19,532 | 3789 | 8 | AAAATAAG | JAGHKT010000009.1 |
| Staphylococcus hominis FDAARGOS_136 | 1,838,641..1,842,445 | 3789 | 8 | AAAATAAG | CP014107 |
| Staphylococcus hominis FDAARGOS_661 | 958,241..962,044 | 3789 | 8 | AAAATAAG | CP054550 |
| Staphylococcus hominis 19A | 2,187,352..2,183,549 | 3789 | 8 | AAAATAAG | CP031277 |
| Staphylococcus hominis C34847 | 1,015,480..1,009,762 | 3789 | 8 | AAAATAAG | CP014567 |
| Staphylococcus epidermidis NW32 | 42,074..45,879 | 3790 | 8 | AAAATAAG | KT726221 |
| Lysinibacillus fusiformis RB-21 | 271,488..267,683 | 3790 | 8 | TATTAAAC | CP010820 |
| Lysinibacillus fusiformis NEB1292 | 1,557,552..1,561,356 | 3791 | 7 | ATTAAAC | CP070490 |
| Lysinibacillus spaericus IAB59 | 4,592,034..4,595,839 | 3790 | 8 | GTTTAATA | CP071741 |
| Salinococcus halodurans H3B36 | 6,428..10,233 2,758,300..2,762,105 | 3790 3790 | 8 8 | TTTAAAAT TTTAAAAT | CP011366 |
| Rothia aeria LPB0401 | 2,602,067..2,605,872 | 3790 | 8 | TTGTTAAG | CP079819 |
| Granulicatella elegans FDAARGOS_1559 | 256,390..260,195 | 3790 | 8 | ATTTTTAT | CP085953 |
| Haemophilus parainfluenzae M1C42_1 | 1,337,751..1,341,555 | 3790 | 8 | AAAATTAT | CP063117 |
| Streptococcus agalactiae 515 | 1,870,022..1,873,812 | 3775 | 8 | TTTAAATT | CP051004 |
| Streptococcus dysgalactiae subsp. equisimilis NCTC7136 | 1,516,598..1,520,403 | 3790 | 8 | AAAAAATC | LS483413 |
| Streptococcus mitis B6 | 838,468..842,273 | 3790 | 8 | TTATTTAT | FN568063 |
| Streptococcus pneumoniae 2245STDY5775520 | 1,015,340..1,019,131 | 3776 | 8 | GAAATATA | LR216027 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Trad, E.I.; Che Hamzah, A.M.; Puah, S.M.; Chua, K.H.; Kwong, S.M.; Yeo, C.C.; Chew, C.H. Comparative Genomic Analysis of a Multidrug-Resistant Staphylococcus hominis ShoR14 Clinical Isolate from Terengganu, Malaysia, Led to the Discovery of Novel Mobile Genetic Elements. Pathogens 2022, 11, 1406. https://doi.org/10.3390/pathogens11121406
Al-Trad EI, Che Hamzah AM, Puah SM, Chua KH, Kwong SM, Yeo CC, Chew CH. Comparative Genomic Analysis of a Multidrug-Resistant Staphylococcus hominis ShoR14 Clinical Isolate from Terengganu, Malaysia, Led to the Discovery of Novel Mobile Genetic Elements. Pathogens. 2022; 11(12):1406. https://doi.org/10.3390/pathogens11121406
Chicago/Turabian StyleAl-Trad, Esra’a I., Ainal Mardziah Che Hamzah, Suat Moi Puah, Kek Heng Chua, Stephen M. Kwong, Chew Chieng Yeo, and Ching Hoong Chew. 2022. "Comparative Genomic Analysis of a Multidrug-Resistant Staphylococcus hominis ShoR14 Clinical Isolate from Terengganu, Malaysia, Led to the Discovery of Novel Mobile Genetic Elements" Pathogens 11, no. 12: 1406. https://doi.org/10.3390/pathogens11121406