


High Throughput Sequencing for Clinical Tuberculosis: An Overview

Abstract
:1. Introduction
2. Whole vs. Targeted Sequencing for TB
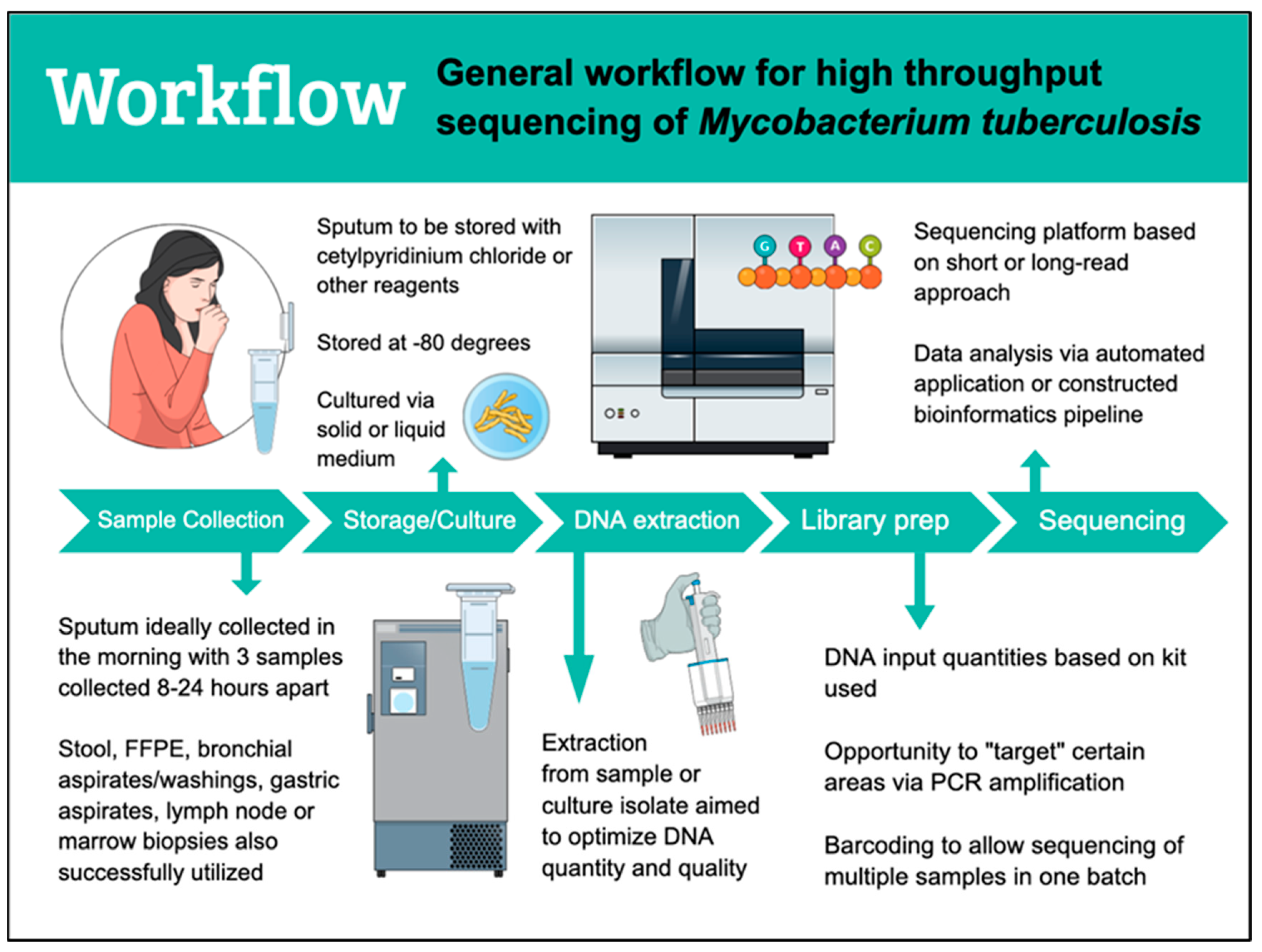
3. Workflow
4. Sequencing Platforms
5. Sequencing Data Analysis and TB Resistance Platforms
6. Future Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. The END TB Strategy; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Nikolenka, A.; Mansjö, M.; Skrahina, A.; Hurevich, H.; Grankov, V.; Nikisins, S.; Dara, M.; Ehsani, S.; Groenheit, R. Whole-genome sequencing differentiates relapse from re-infection in TB. Int. J. Tuberc. Lung Dis. 2021, 25, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.M.; Ip, C.L.; Harrell, R.H.; Evans, J.T.; Kapatai, G.; Dedicoat, M.J.; Eyre, D.W.; Wilson, D.J.; Hawkey, P.M.; Crook, D.W.; et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: A retrospective observational study. Lancet Infect. Dis. 2013, 13, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Köser, C.U.; Bryant, J.M.; Becq, J.; Török, M.E.; Ellington, M.J.; Marti-Renom, M.A.; Carmichael, A.J.; Parkhill, J.; Smith, G.P.; Peacock, S.J. Whole-genome sequencing for rapid susceptibility testing of M. tuberculosis. N. Engl. J. Med. 2013, 369, 290–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohl, T.A.; Diel, R.; Harmsen, D.; Rothgänger, J.; Walter, K.M.; Merker, M.; Weniger, T.; Niemann, S. Whole-genome-based Mycobacterium tuberculosis surveillance: A standardized, portable, and expandable approach. J. Clin. Microbiol. 2014, 52, 2479–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merker, M.; Blin, C.; Mona, S.; Duforet-Frebourg, N.; Lecher, S.; Willery, E.; Blum, M.G.; Rüsch-Gerdes, S.; Mokrousov, I.; Aleksic, E.; et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat. Genet. 2015, 47, 242–249. [Google Scholar] [CrossRef]
- Goig, G.A.; Cancino-Muñoz, I.; Torres-Puente, M.; Villamayor, L.M.; Navarro, D.; Borrás, R.; Comas, I. Whole-genome sequencing of Mycobacterium tuberculosis directly from clinical samples for high-resolution genomic epidemiology and drug resistance surveillance: An observational study. Lancet Microbe 2020, 1, e175–e183. [Google Scholar] [CrossRef]
- Nimmo, C.; Shaw, L.P.; Doyle, R.; Williams, R.; Brien, K.; Burgess, C.; Breuer, J.; Balloux, F.; Pym, A.S. Whole genome sequencing Mycobacterium tuberculosis directly from sputum identifies more genetic diversity than sequencing from culture. BMC Genom. 2019, 20, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, T.M.; Miotto, P.; Köser, C.U.; Fowler, P.W.; Knaggs, J.; Iqbal, Z.; Hunt, M.; Chindelevitch, L.; Farhat, M.; Cirillo, D.M.; et al. The 2021 WHO catalogue of Mycobacterium tuberculosis complex mutations associated with drug resistance: A genotypic analysis. Lancet Microbe 2022, 3, e265–e273. [Google Scholar] [CrossRef]
- Heyckendorf, J.; Andres, S.; Köser, C.U.; Olaru, I.D.; Schön, T.; Sturegård, E.; Beckert, P.; Schleusener, V.; Kohl, T.A.; Hillemann, D.; et al. What Is Resistance? Impact of Phenotypic versus Molecular Drug Resistance Testing on Therapy for Multi- and Extensively Drug-Resistant Tuberculosis. Antimicrob. Agents Chemother. 2018, 62, e01550-17. [Google Scholar] [CrossRef] [Green Version]
- Vogel, M.; Utpatel, C.; Corbett, C.; Kohl, T.A.; Iskakova, A.; Ahmedov, S.; Antonenka, U.; Dreyer, V.; Ibrahimova, A.; Kamarli, C.; et al. Implementation of whole genome sequencing for tuberculosis diagnostics in a low-middle income, high MDR-TB burden country. Sci. Rep. 2021, 11, 15333. [Google Scholar] [CrossRef]
- Meehan, C.J.; Goig, G.A.; Kohl, T.A.; Verboven, L.; Dippenaar, A.; Ezewudo, M.; Farhat, M.R.; Guthrie, J.L.; Laukens, K.; Miotto, P.; et al. Whole genome sequencing of Mycobacterium tuberculosis: Current standards and open issues. Nat. Rev. Microbiol. 2019, 17, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Tafess, K.; Ng, T.T.L.; Lao, H.Y.; Leung, K.S.S.; Tam, K.K.G.; Rajwani, R.; Tam, S.T.Y.; Ho, L.P.K.; Chu, C.M.K.; Gonzalez, D.; et al. Targeted-Sequencing Workflows for Comprehensive Drug Resistance Profiling of Mycobacterium tuberculosis Cultures Using Two Commercial Sequencing Platforms: Comparison of Analytical and Diagnostic Performance, Turnaround Time, and Cost. Clin. Chem. 2020, 66, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.E.; Mace, A.; Seifert, M.; Hetzel, J.; Mshaiel, H.; Suresh, A.; Lemmer, D.; Engelthaler, D.M.; Catanzaro, D.G.; Young, A.G.; et al. Whole-genome and targeted sequencing of drug-resistant Mycobacterium tuberculosis on the iSeq100 and MiSeq: A performance, ease-of-use, and cost evaluation. PLoS Med. 2019, 16, e1002794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, W.S.; Au, C.H.; Chung, Y.; Leung, H.C.M.; Ho, D.N.; Wong, E.Y.L.; Lam, T.W.; Chan, T.L.; Ma, E.S.K.; Tang, B.S.F. Rapid and economical drug resistance profiling with Nanopore MinION for clinical specimens with low bacillary burden of Mycobacterium tuberculosis. BMC Res. Notes 2020, 13, 444. [Google Scholar] [CrossRef] [PubMed]
- Rowneki, M.; Aronson, N.; Du, P.; Sachs, P.; Blakemore, R.; Chakravorty, S.; Levy, S.; Jones, A.L.; Trivedi, G.; Chebore, S.; et al. Detection of drug resistant Mycobacterium tuberculosis by high-throughput sequencing of DNA isolated from acid fast bacilli smears. PLoS ONE 2020, 15, e0232343. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Tu, C.; Chen, W.; Liang, H.; Zhang, W.; Wang, Y.; Jin, Y.; Hu, J.; Sun, Y.; Xu, J.; et al. Rapid Identification of Drug-Resistant Tuberculosis Genes Using Direct PCR Amplification and Oxford Nanopore Technology Sequencing. Can. J. Infect. Dis. Med. Microbiol. 2022, 2022, 7588033. [Google Scholar] [CrossRef]
- Cabibbe, A.M.; Spitaleri, A.; Battaglia, S.; Colman, R.E.; Suresh, A.; Uplekar, S.; Rodwell, T.C.; Cirillo, D.M. Application of Targeted Next-Generation Sequencing Assay on a Portable Sequencing Platform for Culture-Free Detection of Drug-Resistant Tuberculosis from Clinical Samples. J. Clin. Microbiol. 2020, 58, e00632-20. [Google Scholar] [CrossRef]
- Kayomo, M.K.; Mbula, V.N.; Aloni, M.; André, E.; Rigouts, L.; Boutachkourt, F.; de Jong, B.C.; Nkiere, N.M.; Dean, A.S. Targeted next-generation sequencing of sputum for diagnosis of drug-resistant TB: Results of a national survey in Democratic Republic of the Congo. Sci. Rep. 2020, 10, 10786. [Google Scholar] [CrossRef]
- Mesfin, A.B.; Araia, Z.Z.; Beyene, H.N.; Mebrahtu, A.H.; Suud, N.N.; Berhane, Y.M.; Hailu, D.T.; Kassahun, A.Z.; Auguet, O.T.; Dean, A.S.; et al. First molecular-based anti-TB drug resistance survey in Eritrea. Int. J. Tuberc. Lung Dis. 2021, 25, 43–51. [Google Scholar] [CrossRef]
- Sibandze, D.B.; Kay, A.; Dreyer, V.; Sikhondze, W.; Dlamini, Q.; DiNardo, A.; Mtetwa, G.; Lukhele, B.; Vambe, D.; Lange, C.; et al. Rapid molecular diagnostics of tuberculosis resistance by targeted stool sequencing. Genome Med. 2022, 14, 52. [Google Scholar] [CrossRef]
- Colman, R.E.; Anderson, J.; Lemmer, D.; Lehmkuhl, E.; Georghiou, S.B.; Heaton, H.; Wiggins, K.; Gillece, J.D.; Schupp, J.M.; Catanzaro, D.G.; et al. Rapid Drug Susceptibility Testing of Drug-Resistant Mycobacterium tuberculosis Isolates Directly from Clinical Samples by Use of Amplicon Sequencing: A Proof-of-Concept Study. J. Clin. Microbiol. 2016, 54, 2058–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kambli, P.; Ajbani, K.; Kazi, M.; Sadani, M.; Naik, S.; Shetty, A.; Tornheim, J.A.; Singh, H.; Rodrigues, C. Targeted next generation sequencing directly from sputum for comprehensive genetic information on drug resistant Mycobacterium tuberculosis. Tuberculosis 2021, 127, 102051. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Du, W.; Liu, Z.; Che, J.; Li, K.; Che, N. Application of Amplicon-Based Targeted NGS Technology for Diagnosis of Drug-Resistant Tuberculosis Using FFPE Specimens. Microbiol. Spectr. 2022, 10, e0135821. [Google Scholar] [CrossRef] [PubMed]
- Gliddon, H.D.; Frampton, D.; Munsamy, V.; Heaney, J.; Pataillot-Meakin, T.; Nastouli, E.; Pym, A.S.; Steyn, A.J.C.; Pillay, D.; McKendry, R.A. A Rapid Drug Resistance Genotyping Workflow for Mycobacterium tuberculosis, Using Targeted Isothermal Amplification and Nanopore Sequencing. Microbiol. Spectr. 2021, 9, e0061021. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Modongo, C.; Allender, C.; Engelthaler, D.M.; Warren, R.M.; Zetola, N.M.; Shin, S.S. Utility of Targeted, Amplicon-Based Deep Sequencing to Detect Resistance to First-Line Tuberculosis Drugs in Botswana. Antimicrob. Agents Chemother. 2019, 63, e00982-19. [Google Scholar] [CrossRef] [PubMed]
- Mariner-Llicer, C.; Goig, G.A.; Zaragoza-Infante, L.; Torres-Puente, M.; Villamayor, L.; Navarro, D.; Borras, R.; Chiner-Oms, Á.; Comas, I. Accuracy of an amplicon-sequencing nanopore approach to identify variants in tuberculosis drug-resistance-associated genes. Microb. Genom. 2021, 7, 000740. [Google Scholar] [CrossRef] [PubMed]
- Tagliani, E.; Hassan, M.O.; Waberi, Y.; De Filippo, M.R.; Falzon, D.; Dean, A.; Zignol, M.; Supply, P.; Abdoulkader, M.A.; Hassangue, H.; et al. Culture and Next-generation sequencing-based drug susceptibility testing unveil high levels of drug-resistant-TB in Djibouti: Results from the first national survey. Sci. Rep. 2017, 7, 17672. [Google Scholar] [CrossRef] [Green Version]
- Colman, R.E.; Schupp, J.M.; Hicks, N.D.; Smith, D.E.; Buchhagen, J.L.; Valafar, F.; Crudu, V.; Romancenco, E.; Noroc, E.; Jackson, L.; et al. Detection of Low-Level Mixed-Population Drug Resistance in Mycobacterium tuberculosis Using High Fidelity Amplicon Sequencing. PLoS ONE 2015, 10, e0126626. [Google Scholar] [CrossRef] [Green Version]
- Jouet, A.; Gaudin, C.; Badalato, N.; Allix-Béguec, C.; Duthoy, S.; Ferré, A.; Diels, M.; Laurent, Y.; Contreras, S.; Feuerriegel, S.; et al. Deep amplicon sequencing for culture-free prediction of susceptibility or resistance to 13 anti-tuberculous drugs. Eur. Respir. J. 2021, 57, 2002338. [Google Scholar] [CrossRef]
- WHO Guidelines Approved by the Guidelines Review Committee. In Guidance for National Tuberculosis Programmes on the Management of Tuberculosis in Children; World Health Organization: Geneva, Switzerland, 2014.
- Hiza, H.; Doulla, B.; Sasamalo, M.; Hella, J.; Kamwela, L.; Mhimbira, F.; Reither, K.; Gagneux, S.; Jugheli, L.; Fenner, L. Preservation of sputum samples with cetylpyridinium chloride (CPC) for tuberculosis cultures and Xpert MTB/RIF in a low-income country. BMC Infect. Dis. 2017, 17, 542. [Google Scholar] [CrossRef]
- Sanoussi, C.N.; de Jong, B.C.; Affolabi, D.; Meehan, C.J.; Odoun, M.; Rigouts, L. Storage of Sputum in Cetylpyridinium Chloride, OMNIgene.SPUTUM, and Ethanol Is Compatible with Molecular Tuberculosis Diagnostic Testing. J. Clin. Microbiol. 2019, 57, e00275-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Votintseva, A.A.; Pankhurst, L.J.; Anson, L.W.; Morgan, M.R.; Gascoyne-Binzi, D.; Walker, T.M.; Quan, T.P.; Wyllie, D.H.; Del Ojo Elias, C.; Wilcox, M.; et al. Mycobacterial DNA extraction for whole-genome sequencing from early positive liquid (MGIT) cultures. J. Clin. Microbiol. 2015, 53, 1137–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuerriegel, S.; Kohl, T.A.; Utpatel, C.; Andres, S.; Maurer, F.P.; Heyckendorf, J.; Jouet, A.; Badalato, N.; Foray, L.; Fouad Kamara, R.; et al. Rapid genomic first- and second-line drug resistance prediction from clinical Mycobacterium tuberculosis specimens using Deeplex-MycTB. Eur. Respir. J. 2021, 57, 2001796. [Google Scholar] [CrossRef]
- Hess, J.F.; Kohl, T.A.; Kotrová, M.; Rönsch, K.; Paprotka, T.; Mohr, V.; Hutzenlaub, T.; Brüggemann, M.; Zengerle, R.; Niemann, S.; et al. Library preparation for next generation sequencing: A review of automation strategies. Biotechnol. Adv. 2020, 41, 107537. [Google Scholar] [CrossRef] [PubMed]
- WHO. The Use of Next-Generation Sequencing Technologies for the Detection of Mutations Associated with Drug Resistance in Mycobacterium Tuberculosis Complex: Technical Guide; Report No.: Contract No.: WHO/CDS/TB/2018.19; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Modlin, S.J.; Robinhold, C.; Morrissey, C.; Mitchell, S.N.; Ramirez-Busby, S.M.; Shmaya, T.; Valafar, F. Exact mapping of Illumina blind spots in the Mycobacterium tuberculosis genome reveals platform-wide and workflow-specific biases. Microb. Genom. 2021, 7, 000465. [Google Scholar] [CrossRef] [PubMed]
- Daum, L.T.; Rodriguez, J.D.; Worthy, S.A.; Ismail, N.A.; Omar, S.V.; Dreyer, A.W.; Fourie, P.B.; Hoosen, A.A.; Chambers, J.P.; Fischer, G.W. Next-generation ion torrent sequencing of drug resistance mutations in Mycobacterium tuberculosis strains. J. Clin. Microbiol. 2012, 50, 3831–3837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavel, I.; Macovei, I.I.; Péter, S.; Diculencu, D.; Rusu-Cordunean, F.; Grigoriu, B.D. Detection of first and second line drug resistance mutations from multi drug resistant Mycobacterium tuberculosis strains by Ion Torrent whole genome sequencing (WGS). Eur. Respir. J. 2016, 48, PA1910. [Google Scholar] [CrossRef]
- Lee, R.S.; Proulx, J.F.; McIntosh, F.; Behr, M.A.; Hanage, W.P. Previously undetected super-spreading of Mycobacterium tuberculosis revealed by deep sequencing. eLife 2020, 9, e53245. [Google Scholar] [CrossRef]
- Ley, S.D.; de Vos, M.; Van Rie, A.; Warren, R.M. Deciphering Within-Host Microevolution of Mycobacterium tuberculosis through Whole-Genome Sequencing: The Phenotypic Impact and Way Forward. Microbiol. Mol. Biol. Rev. 2019, 83, e00062-18. [Google Scholar] [CrossRef] [Green Version]
- ONT. Accuracy. Oxford Nanopore Technologies. 2022. Available online: https://nanoporetech.com/accuracy (accessed on 15 August 2022).
- Smith, C.; Halse, T.A.; Shea, J.; Modestil, H.; Fowler, R.C.; Musser, K.A.; Escuyer, V.; Lapierre, P. Assessing Nanopore Sequencing for Clinical Diagnostics: A Comparison of Next-Generation Sequencing (NGS) Methods for Mycobacterium tuberculosis. J. Clin. Microbiol. 2020, 59, e00583-20. [Google Scholar] [CrossRef]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ONT. Oxford Nanopore Announces Positive Evaluation of New Method for Rapid Drug-Resistant Tuberculosis (DR-TB) Profiling. Oxord Nanopore Technologies. Updated 23 March 2022. Available online: https://nanoporetech.com/about-us/news/oxford-nanopore-announces-positive-evaluation-new-method-rapid-drug-resistant (accessed on 15 September 2022).
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- GitHub. vSNP. GitHub. 2020. Available online: https://github.com/USDA-VS/vSNP (accessed on 12 September 2022).
- GitHub. SNiPgenie. Available online: https://github.com/dmnfarrell/snipgenie2020 (accessed on 15 September 2022).
- Kohl, T.A.; Utpatel, C.; Schleusener, V.; De Filippo, M.R.; Beckert, P.; Cirillo, D.M.; Niemann, S. MTBseq: A comprehensive pipeline for whole genome sequence analysis of Mycobacterium tuberculosis complex isolates. PeerJ 2018, 6, e5895. [Google Scholar] [CrossRef] [Green Version]
- GitHub. BovTB. Available online: https://github.com/oxfordmmm/BovTB-nf-docker2019 (accessed on 15 September 2022).
- Steiner, A.; Stucki, D.; Coscolla, M.; Borrell, S.; Gagneux, S. KvarQ: Targeted and direct variant calling from fastq reads of bacterial genomes. BMC Genom. 2014, 15, 881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuerriegel, S.; Schleusener, V.; Beckert, P.; Kohl, T.A.; Miotto, P.; Cirillo, D.M.; Cabibbe, A.M.; Niemann, S.; Fellenberg, K. PhyResSE: A Web Tool Delineating Mycobacterium tuberculosis Antibiotic Resistance and Lineage from Whole-Genome Sequencing Data. J. Clin. Microbiol. 2015, 53, 1908–1914. [Google Scholar] [CrossRef] [Green Version]
- Hunt, M.; Bradley, P.; Lapierre, S.G.; Heys, S.; Thomsit, M.; Hall, M.B.; Malone, K.M.; Wintringer, P.; Walker, T.M.; Cirillo, D.M.; et al. Antibiotic resistance prediction for Mycobacterium tuberculosis from genome sequence data with Mykrobe. Wellcome Open Res. 2019, 4, 191. [Google Scholar] [CrossRef]


{kind=link}
| Author/Year | Location | tHTS Kit | Index Test Sample | Platform | Reference Standard/Comparisons |
|---|---|---|---|---|---|
| Cabbibe 2020 [18] | Italy | Deeplex Myc-TB | Sputum | MinION | Deeplex Myc-TB amplicons on MiniSeq |
| Kambli 2021 [23] | Mumbai, India | Deeplex Myc-TB | Sputum, isolates | iSeq | pDST from MGIT; LPA; pyrosequencing |
| Kayomo 2020 [19] | Congo | Deeplex Myc-TB | Sputum | MiSeq | Xpert MTB/RIF |
| Colman 2019 [14] | United States | SMOR assay [24] | Isolates | MiSeq, iSeq | WGS (MiSeq and iSeq) |
| Tafess 2020 [13] | Hong Kong, Ethiopia | In-house PCR (19 gene targets) | Isolates | MiSeq, MinION | pDST from MGIT |
| Gliddon 2021 [25] | Kwazulu, South Africa | In-house RPA (isothermal) | Isolates | MinION | WGS (HiSeq), pDST from MGIT and solid agar |
| Sibandze 2022 [21] | Eswatini, Germany | Deeplex Myc-TB | Stool | NextSeq | pDST from MGIT |
| Wang 2019 [26] | Botswana | SMOR assay [24] | Isolates | MiSeq | pDST on MGIT; LPA, Xpert MTB/RIF |
| Mariner-Llicer 2021 [27] | Spain | In-house assay (11 gene targets) | Sputum, isolates | MinION | WGS (MiSeq) |
| Mesfin 2021 [20] | Eritrea | Deeplex Myc-TB | Sputum | MiniSeq | Xpert MTB/RIF |
| Tagliani 2017 [28] | Djibouti | Deeplex Myc-TB | Sputum | MiniSeq | WGS (HiSeq) from isolates |
| Song 2022 [24] | China | In house PCR (11 gene targets) | FFPE tissues | Ion Proton | pDST from microtitre plate |
| Chan 2020 [15] | Hong Kong | In-house PCR (10 gene targets) | Bronchial aspirate, LN, sputum, bone marrow | MinION, MiSeq | pDST from MGIT; LPA |
| Rowneki 2020 [16] | Ghana, Kenya, Uganda, Zambia | In house PCR (17 gene targets) | Sputum | MiSeq | Sanger sequencing on subset of samples |
| Colman 2016 [22] | Moldova | SMOR assay | Sputum | MiSeq | pDST from MGIT |
| Colman 2015 [29] | Moldova | SMOR assay | Sputum | MiSeq | pDST from MGIT |
| Zhao 2022 [17] | Shanghai, China | In-house PCR (7 gene targets) | Sputum | GridION | Sanger sequencing, pDST |
| Jouet 2021 [30] | Djibouti, Congo | Deeplex Myc-TB | Sputum | MiSeq | WGS, pDST from Löwenstein–Jensen or Middlebrook 7H11 agar |
| Platforms | Characteristics | Pros | Cons | Studies |
|---|---|---|---|---|
| Illumina iSeq MiniSeq Miseq Nextseq HiSeq NovaSeq | Short-read (2 × 150 bp, MISEQ 2 × 300 bp) Run time 4–72 h | Low error rate (99.9% accuracy) Platforms vary in low to high throughput | Difficulty in sequencing repetitive regions [38] Per base error rate increases with read length (trimming can improve) Long run times | Sibandze 2021 [21]; Kambli 2021 [23]; Kayomo 2020 [19]; Wang 2019 [26]; Colman 2019 [14]; Mesfin 2021 [20]; Tagliani 2017 [28]; Vogel 2021 [11] |
| Thermo fisher Ion torrent Proton PGM S5 | Short-read (200–400 bp) Run time 3–24 h | Short run time Low error rate | Low performance on homopolymer regions High cost per sample | Daum 2012 [39]; Pavel 2016 [40] |
| Pacbio RSII Sequel | Long-read (10–60 kb) Run time up to 20 h | Short run time, long read length | High cost, high error rate (single base pair deletions most common, can improve with increased depth) | Lee 2019 [41]; Ley 2019 [42] |
| Oxford Nanopore Minion Gridion Promethion | Long-read (900 kb +) Run time 30 min to 48 h | Short run time, long read length Increasing portability | Historically higher error rate (>98% reported accuracy with newer technology) [43,44] | Gliddon 2021 [25]; Mariner-Llicer 2021 [27]; Zhao 2022 [17] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ness, T.E.; DiNardo, A.; Farhat, M.R. High Throughput Sequencing for Clinical Tuberculosis: An Overview. Pathogens 2022, 11, 1343. https://doi.org/10.3390/pathogens11111343
Ness TE, DiNardo A, Farhat MR. High Throughput Sequencing for Clinical Tuberculosis: An Overview. Pathogens. 2022; 11(11):1343. https://doi.org/10.3390/pathogens11111343
Chicago/Turabian StyleNess, Tara E., Andrew DiNardo, and Maha R. Farhat. 2022. "High Throughput Sequencing for Clinical Tuberculosis: An Overview" Pathogens 11, no. 11: 1343. https://doi.org/10.3390/pathogens11111343