
Regulatory Role of Phospholipids in Hepatitis C Virus Replication and Protein Function


Abstract
:1. Introduction
2. HCV Genome Replication
2.1. HCV Structural and Nonstructural Proteins
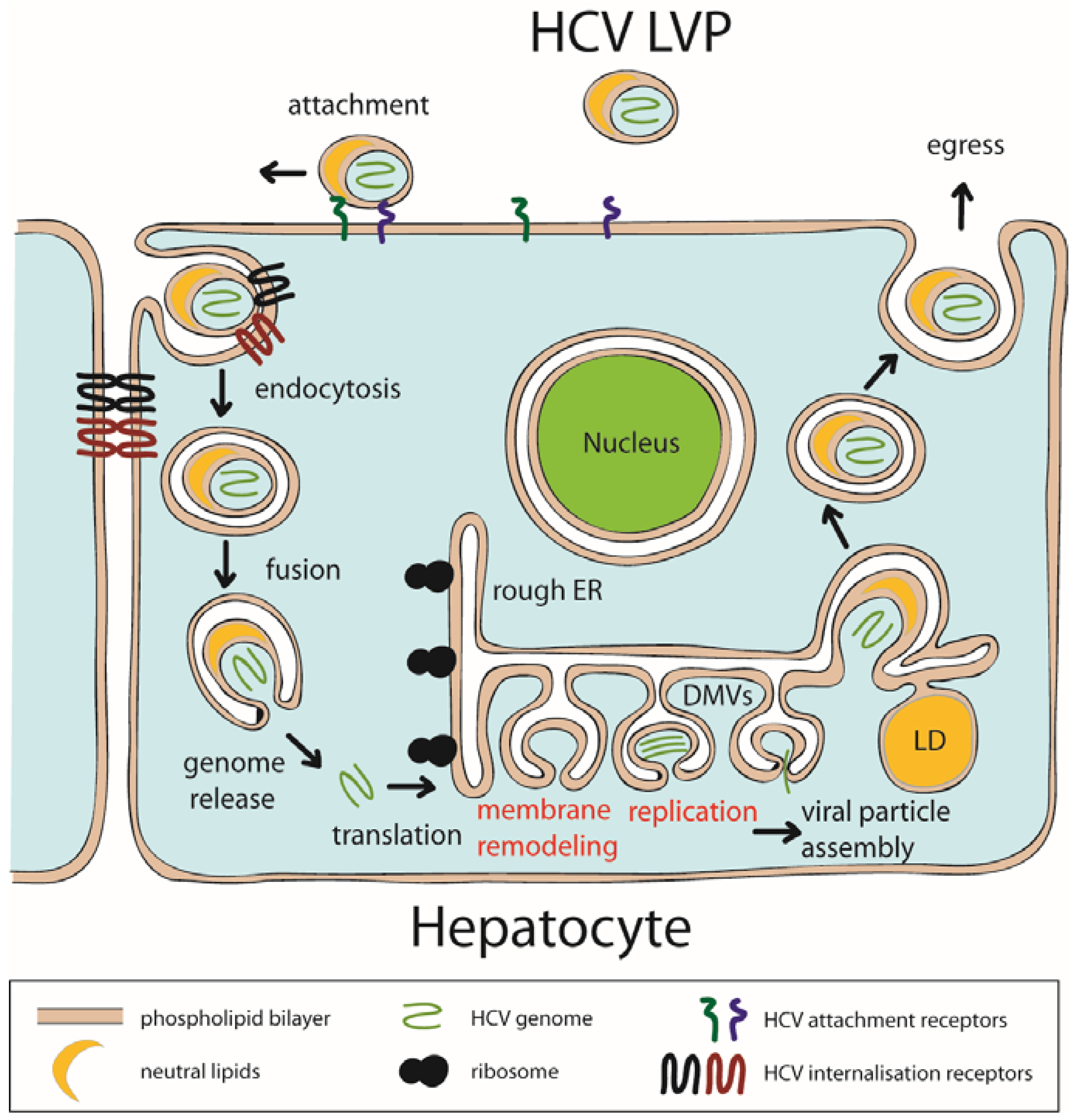
2.2. HCV Replication Organelle
2.3. Role of Phospholipids in HCV Genome Replication
2.3.1. Direct Interaction of NS Proteins with Phospholipids and Stimulation of Replication
2.3.2. Indirect Effects of Phospholipids on Replication Complex Assembly
3. Reprogramming/Hijacking Host PI4KIIIα
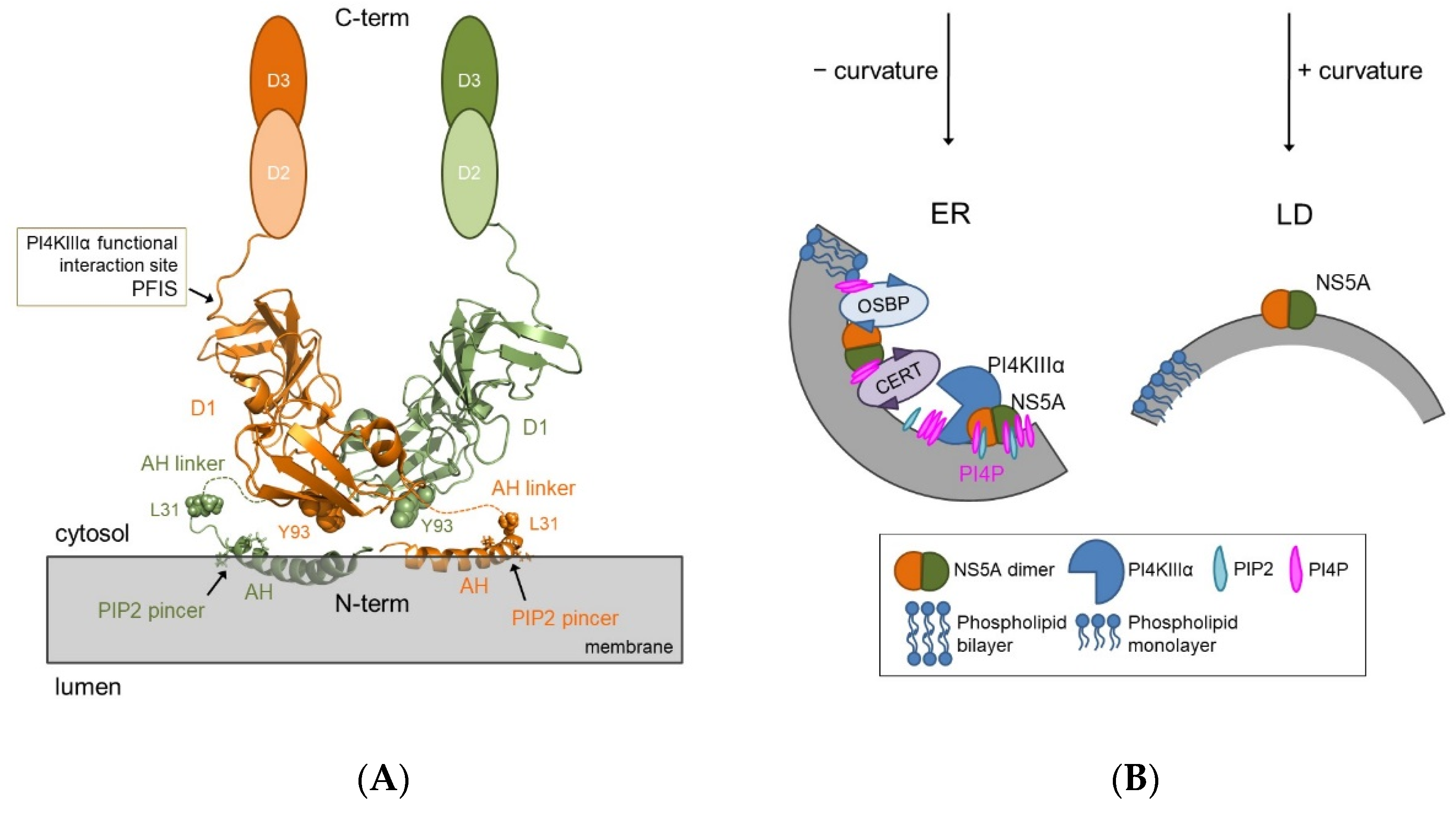
4. Structure-Function Relationship of NS5A and Role of the Lipid Environment
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| aa | amino acid |
| AH | amphipathic helix |
| BAAPP | basic amino acid PIP2 pincer |
| BMRB | Biological Magnetic Resonance Data Bank |
| CERT | ceramide transfer protein; DFCP1, double FYVE-containing protein 1 |
| DMV | double membrane vesicles; DRM, detergent-resistant membrane |
| FAPP2 | four-phosphate adaptor protein |
| HCV | hepatitis C virus |
| IRES | internal ribosome entry site |
| LD | lipid droplet |
| LVP | lipo-viro-particle |
| MMV | multimembrane vesicles |
| MW | membranous web |
| NCP | negatively charged phospholipid |
| NMR | nuclear magnetic resonance |
| NS | nonstructural |
| NS5Ai | NS5A inhibitor |
| ORF | open reading frame |
| OSBP | oxysterol-binding protein |
| PDB | Protein Data Bank |
| PI | phosphatidylinositol |
| PI3P | phosphatidylinositol 3-phosphate |
| PI4P | phosphatidylinositol 4-phosphate |
| PIP2 | phosphatidylinositol (4,5)-bisphosphate |
| PIP3 | phosphatidylinositol (3,4,5)-trisphosphate |
| PI4KIIIα | phosphatidylinositol 4-kinase III alpha |
| RNA | ribonucleic acid |
| siRNA | small interfering ribonucleic acid |
| SM | sphingomyelin |
| SREBP | sterol regulatory element-binding protein |
| UTR | untranslated region |
References
- Zhang, J.; Lan, Y.; Sanyal, S. Modulation of Lipid Droplet Metabolism—A Potential Target for Therapeutic Intervention in Flaviviridae Infections. Front. Microbiol. 2017, 8, 2286. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring Cellular Networks by Members of the Flaviviridae Family. Nat. Rev. Microbiol. 2018, 16, 125–142. [Google Scholar] [CrossRef]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the Hepatitis C Virus RNA Replication Complex in Huh-7 Cells Harboring Subgenomic Replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar] [CrossRef] [Green Version]
- Paul, D.; Hoppe, S.; Saher, G.; Krijnse-Locker, J.; Bartenschlager, R. Morphological and Biochemical Characterization of the Membranous Hepatitis C Virus Replication Compartment. J. Virol. 2013, 87, 10612–10627. [Google Scholar] [CrossRef] [Green Version]
- Romero-Brey, I.; Berger, C.; Kallis, S.; Kolovou, A.; Paul, D.; Lohmann, V.; Bartenschlager, R. NS5A Domain 1 and Polyprotein Cleavage Kinetics Are Critical for Induction of Double-Membrane Vesicles Associated with Hepatitis C Virus Replication. mBio 2015, 6, e00759. [Google Scholar] [CrossRef] [Green Version]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.-A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal Proteome and Lipidome Profiles Reveal Hepatitis C Virus-Associated Reprogramming of Hepatocellular Metabolism and Bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, S.; Krajewski, M.; Scherer, C.; Scholz, V.; Mordhorst, V.; Truschow, P.; Schöbel, A.; Reimer, R.; Schwudke, D.; Herker, E. Complex Lipid Metabolic Remodeling Is Required for Efficient Hepatitis C Virus Replication. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1041–1056. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Lohmann, V.; Penin, F. The Molecular and Structural Basis of Advanced Antiviral Therapy for Hepatitis C Virus Infection. Nat. Rev. Microbiol. 2013, 11, 482–496. [Google Scholar] [CrossRef] [Green Version]
- Moradpour, D.; Penin, F. Hepatitis C Virus Proteins: From Structure to Function. Curr. Top. Microbiol. Immunol. 2013, 369, 113–142. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Körner, F.; Koch, J.-O.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of Subgenomic Hepatitis C Virus RNAs in a Hepatoma Cell Line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef] [Green Version]
- Niepmann, M.; Gerresheim, G.K. Hepatitis C Virus Translation Regulation. Int. J. Mol. Sci. 2020, 21, 2328. [Google Scholar] [CrossRef] [Green Version]
- Dultz, G.; Shimakami, T.; Schneider, M.; Murai, K.; Yamane, D.; Marion, A.; Zeitler, T.M.; Stross, C.; Grimm, C.; Richter, R.M.; et al. Extended Interaction Networks with HCV Protease NS3–4A Substrates Explain the Lack of Adaptive Capability against Protease Inhibitors. J. Biol. Chem. 2020, 295, 13862–13874. [Google Scholar] [CrossRef] [PubMed]
- Shimakami, T.; Welsch, C.; Yamane, D.; McGivern, D.R.; Yi, M.; Zeuzem, S.; Lemon, S.M. Protease Inhibitor-Resistant Hepatitis C Virus Mutants with Reduced Fitness from Impaired Production of Infectious Virus. Gastroenterology 2011, 140, 667–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsch, C.; Shimakami, T.; Hartmann, C.; Yang, Y.; Domingues, F.S.; Lengauer, T.; Zeuzem, S.; Lemon, S.M. Peptidomimetic Escape Mechanisms Arise via Genetic Diversity in the Ligand-Binding Site of the Hepatitis C Virus NS3/4A Serine Protease. Gastroenterology 2012, 142, 654–663. [Google Scholar] [CrossRef] [Green Version]
- Doncheva, N.T.; Domingues, F.S.; McGivern, D.R.; Shimakami, T.; Zeuzem, S.; Lengauer, T.; Lange, C.M.; Albrecht, M.; Welsch, C. Near-Neighbor Interactions in the NS3–4A Protease of HCV Impact Replicative Fitness of Drug-Resistant Viral Variants. J. Mol. Biol. 2019, 431, 2354–2368. [Google Scholar] [CrossRef] [PubMed]
- Egger, D.; Wölk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of Hepatitis C Virus Proteins Induces Distinct Membrane Alterations Including a Candidate Viral Replication Complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef] [Green Version]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.-Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-Dimensional Architecture and Biogenesis of Membrane Structures Associated with Hepatitis C Virus Replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef] [Green Version]
- Kandangwa, M.; Liu, Q. HCV NS5A Hyperphosphorylation Is Involved in Viral Translation Modulation. Biochem. Biophys. Res. Commun. 2019, 520, 192–197. [Google Scholar] [CrossRef]
- Shanmugam, S.; Nichols, A.K.; Saravanabalaji, D.; Welsch, C.; Yi, M. HCV NS5A Dimer Interface Residues Regulate HCV Replication by Controlling Its Self-Interaction, Hyperphosphorylation, Subcellular Localization and Interaction with Cyclophilin, A. PLoS Pathog. 2018, 14, e1007177. [Google Scholar] [CrossRef] [Green Version]
- Pawlotsky, J.-M.; Negro, F.; Aghemo, A.; Berenguer, M.; Dalgard, O.; Dusheiko, G.; Marra, F.; Puoti, M.; Wedemeyer, H. EASL Recommendations on Treatment of Hepatitis C 2018. J. Hepatol. 2018, 69, 461–511. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, C.; Singh, M.; Das, D.; Chaudhuri, S.; Mukhopadhyay, A. Current Therapeutics against HCV. Virusdisease 2021, 32, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Cotter, T.G.; Jensen, D.M. Glecaprevir/Pibrentasvir for the Treatment of Chronic Hepatitis C: Design, Development and Place in Therapy. Drug Des. Dev. Ther. 2019, 13, 2565–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarrazin, C.; Zimmermann, T.; Berg, T.; Hinrichsen, H.; Mauss, S.; Wedemeyer, H.; Zeuzem, S.; Deutsche Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselkrankheiten (DGVS); Deutsche Gesellschaft für Pathologie e.V. (DGP) und Bundesverband Deutscher Pathologen (BDP); Deutsche Leberstiftung; et al. Prophylaxe, Diagnostik und Therapie der Hepatitis-C-Virus(HCV)-Infektion. Z. Gastroenterol. 2020, 58, 1110–1131. [Google Scholar] [CrossRef]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.-H.; O’Boyle, D.R.; et al. Chemical Genetics Strategy Identifies an HCV NS5A Inhibitor with a Potent Clinical Effect. Nature 2010, 465, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Foster, G.R. Second Generation Direct-Acting Antivirals—Do We Expect Major Improvements? J. Hepatol. 2016, 65, S130–S142. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.-H.; O’Boyle, D.R.; Fridell, R.A.; Langley, D.R.; Wang, C.; Roberts, S.B.; Nower, P.; Johnson, B.M.; Moulin, F.; Nophsker, M.J.; et al. Resensitizing Daclatasvir-Resistant Hepatitis C Variants by Allosteric Modulation of NS5A. Nature 2015, 527, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.Y.; Ray, A.S. HCV RdRp, Sofosbuvir and Beyond. Enzymes 2021, 49, 63–82. [Google Scholar] [CrossRef]
- Wolff, G.; Melia, C.E.; Snijder, E.J.; Bárcena, M. Double-Membrane Vesicles as Platforms for Viral Replication. Trends Microbiol. 2020, 28, 1022–1033. [Google Scholar] [CrossRef]
- Ferraris, P.; Blanchard, E.; Roingeard, P. Ultrastructural and Biochemical Analyses of Hepatitis C Virus-Associated Host Cell Membranes. J. Gen. Virol. 2010, 91, 2230–2237. [Google Scholar] [CrossRef]
- Ferraris, P.; Beaumont, E.; Uzbekov, R.; Brand, D.; Gaillard, J.; Blanchard, E.; Roingeard, P. Sequential Biogenesis of Host Cell Membrane Rearrangements Induced by Hepatitis C Virus Infection. Cell. Mol. Life Sci. CMLS 2013, 70, 1297–1306. [Google Scholar] [CrossRef] [Green Version]
- Aizaki, H.; Lee, K.-J.; Sung, V.M.-H.; Ishiko, H.; Lai, M.M.C. Characterization of the Hepatitis C Virus RNA Replication Complex Associated with Lipid Rafts. Virology 2004, 324, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Quinkert, D.; Bartenschlager, R.; Lohmann, V. Quantitative Analysis of the Hepatitis C Virus Replication Complex. J. Virol. 2005, 79, 13594–13605. [Google Scholar] [CrossRef] [Green Version]
- Miyanari, Y.; Hijikata, M.; Yamaji, M.; Hosaka, M.; Takahashi, H.; Shimotohno, K. Hepatitis C Virus Non-Structural Proteins in the Probable Membranous Compartment Function in Viral Genome Replication. J. Biol. Chem. 2003, 278, 50301–50308. [Google Scholar] [CrossRef] [Green Version]
- Scutigliani, E.M.; Kikkert, M. Interaction of the Innate Immune System with Positive-Strand RNA Virus Replication Organelles. Cytokine Growth Factor Rev. 2017, 37, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Madan, V.; Bartenschlager, R. Hepatitis C Virus RNA Replication and Assembly: Living on the Fat of the Land. Cell Host Microbe 2014, 16, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyre, N.S.; Fiches, G.N.; Aloia, A.L.; Helbig, K.J.; McCartney, E.M.; McErlean, C.S.P.; Li, K.; Aggarwal, A.; Turville, S.G.; Beard, M.R. Dynamic Imaging of the Hepatitis C Virus NS5A Protein during a Productive Infection. J. Virol. 2014, 88, 3636–3652. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Tai, A.W. Continuous de Novo Generation of Spatially Segregated Hepatitis C Virus Replication Organelles Revealed by Pulse-Chase Imaging. J. Hepatol. 2017, 66, 55–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, S.; Rebhan, I.; Backes, P.; Romero-Brey, I.; Erfle, H.; Matula, P.; Kaderali, L.; Poenisch, M.; Blankenburg, H.; Hiet, M.-S.; et al. Recruitment and Activation of a Lipid Kinase by Hepatitis C Virus NS5A Is Essential for Integrity of the Membranous Replication Compartment. Cell Host Microbe 2011, 9, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Tai, A.W.; Salloum, S. The Role of the Phosphatidylinositol 4-Kinase PI4KA in Hepatitis C Virus-Induced Host Membrane Rearrangement. PLoS ONE 2011, 6, e26300. [Google Scholar] [CrossRef] [PubMed]
- Lyn, R.K.; Singaravelu, R.; Kargman, S.; O’Hara, S.; Chan, H.; Oballa, R.; Huang, Z.; Jones, D.M.; Ridsdale, A.; Russell, R.S.; et al. Stearoyl-CoA Desaturase Inhibition Blocks Formation of Hepatitis C Virus-Induced Specialized Membranes. Sci. Rep. 2014, 4, 4549. [Google Scholar] [CrossRef] [Green Version]
- Palomares-Jerez, M.F.; Nemesio, H.; Franquelim, H.G.; Castanho, M.A.R.B.; Villalaín, J. N-Terminal AH2 Segment of Protein NS4B from Hepatitis C Virus. Binding to and Interaction with Model Biomembranes. Biochim. Biophys. Acta 2013, 1828, 1938–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashworth Briggs, E.L.; Gomes, R.G.B.; Elhussein, M.; Collier, W.; Findlow, I.S.; Khalid, S.; McCormick, C.J.; Williamson, P.T.F. Interaction between the NS4B Amphipathic Helix, AH2, and Charged Lipid Headgroups Alters Membrane Morphology and AH2 Oligomeric State--Implications for the Hepatitis C Virus Life Cycle. Biochim. Biophys. Acta 2015, 1848, 1671–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, N.-J.; Lee, C.; Pang, P.S.; Pham, E.A.; Fram, B.; Nguyen, K.; Xiong, A.; Sklan, E.H.; Elazar, M.; Koytak, E.S.; et al. Phosphatidylinositol 4,5-Bisphosphate Is an HCV NS5A Ligand and Mediates Replication of the Viral Genome. Gastroenterology 2015, 148, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-O.; Jackman, J.A.; Elazar, M.; Cho, S.-J.; Glenn, J.S.; Cho, N.-J. Quantitative Evaluation of Viral Protein Binding to Phosphoinositide Receptors and Pharmacological Inhibition. Anal. Chem. 2017, 89, 9742–9750. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, H.; Okamoto, K.; Aoki, M.; Kato, H.; Katsume, A.; Ohta, A.; Tsukuda, T.; Shimma, N.; Aoki, Y.; Arisawa, M.; et al. Host Sphingolipid Biosynthesis as a Target for Hepatitis C Virus Therapy. Nat. Chem. Biol. 2005, 1, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Gouttenoire, J.; Montserret, R.; Paul, D.; Castillo, R.; Meister, S.; Bartenschlager, R.; Penin, F.; Moradpour, D. Aminoterminal Amphipathic α-Helix AH1 of Hepatitis C Virus Nonstructural Protein 4B Possesses a Dual Role in RNA Replication and Virus Production. PLoS Pathog. 2014, 10, e1004501. [Google Scholar] [CrossRef]
- Weng, L.; Hirata, Y.; Arai, M.; Kohara, M.; Wakita, T.; Watashi, K.; Shimotohno, K.; He, Y.; Zhong, J.; Toyoda, T. Sphingomyelin Activates Hepatitis C Virus RNA Polymerase in a Genotype-Specific Manner. J. Virol. 2010, 84, 11761–11770. [Google Scholar] [CrossRef] [Green Version]
- Hirata, Y.; Ikeda, K.; Sudoh, M.; Tokunaga, Y.; Suzuki, A.; Weng, L.; Ohta, M.; Tobita, Y.; Okano, K.; Ozeki, K.; et al. Self-Enhancement of Hepatitis C Virus Replication by Promotion of Specific Sphingolipid Biosynthesis. PLoS Pathog. 2012, 8, e1002860. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Pontremoli, C.; Pozzoli, U.; Vertemara, J.; De Gioia, L.; Clerici, M.; Sironi, M. Evolutionary Analysis Provides Insight into the Origin and Adaptation of HCV. Front. Microbiol. 2018, 9, 854. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.T.; Lee, K.-J.; Aizaki, H.; Hwang, S.B.; Lai, M.M.C. Hepatitis C Virus RNA Replication Occurs on a Detergent-Resistant Membrane That Cofractionates with Caveolin-2. J. Virol. 2003, 77, 4160–4168. [Google Scholar] [CrossRef] [Green Version]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The Mystery of Membrane Organization: Composition, Regulation and Roles of Lipid Rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gewaid, H.; Aoyagi, H.; Arita, M.; Watashi, K.; Suzuki, R.; Sakai, S.; Kumagai, K.; Yamaji, T.; Fukasawa, M.; Kato, F.; et al. Sphingomyelin Is Essential for the Structure and Function of the Double-Membrane Vesicles in Hepatitis C Virus RNA Replication Factories. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Wang, H.; Perry, J.W.; Lauring, A.S.; Neddermann, P.; De Francesco, R.; Tai, A.W. Oxysterol-Binding Protein Is a Phosphatidylinositol 4-Kinase Effector Required for HCV Replication Membrane Integrity and Cholesterol Trafficking. Gastroenterology 2014, 146, 1373.e1-11–1385.e1-11. [Google Scholar] [CrossRef]
- Antonny, B.; Bigay, J.; Mesmin, B. The Oxysterol-Binding Protein Cycle: Burning Off PI(4)P to Transport Cholesterol. Annu. Rev. Biochem. 2018, 87, 809–837. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Katikaneni, D.S.; Han, Q.; Sanchez-Felipe, L.; Hanada, K.; Ambrose, R.L.; Mackenzie, J.M.; Konan, K.V. Modulation of Hepatitis C Virus Genome Replication by Glycosphingolipids and Four-Phosphate Adaptor Protein 2. J. Virol. 2014, 88, 12276–12295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delang, L.; Harak, C.; Benkheil, M.; Khan, H.; Leyssen, P.; Andrews, M.; Lohmann, V.; Neyts, J. PI4KIII Inhibitor Enviroxime Impedes the Replication of the Hepatitis C Virus by Inhibiting PI3 Kinases. J. Antimicrob. Chemother. 2018, 73, 3375–3384. [Google Scholar] [CrossRef]
- Twu, W.-I.; Lee, J.-Y.; Kim, H.; Prasad, V.; Cerikan, B.; Haselmann, U.; Tabata, K.; Bartenschlager, R. Contribution of Autophagy Machinery Factors to HCV and SARS-CoV-2 Replication Organelle Formation. Cell Rep. 2021, 37, 110049. [Google Scholar] [CrossRef]
- Mohl, B.-P.; Bartlett, C.; Mankouri, J.; Harris, M. Early Events in the Generation of Autophagosomes Are Required for the Formation of Membrane Structures Involved in Hepatitis C Virus Genome Replication. J. Gen. Virol. 2016, 97, 680–693. [Google Scholar] [CrossRef] [Green Version]
- Bochkov, V.N.; Oskolkova, O.V.; Birukov, K.G.; Levonen, A.-L.; Binder, C.J.; Stöckl, J. Generation and Biological Activities of Oxidized Phospholipids. Antioxid. Redox Signal. 2010, 12, 1009–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzimenti, S.; Ciamporcero, E.; Daga, M.; Pettazzoni, P.; Arcaro, A.; Cetrangolo, G.; Minelli, R.; Dianzani, C.; Lepore, A.; Gentile, F.; et al. Interaction of Aldehydes Derived from Lipid Peroxidation and Membrane Proteins. Front. Physiol. 2013, 4, 242. [Google Scholar] [CrossRef] [Green Version]
- Yamane, D.; McGivern, D.R.; Wauthier, E.; Yi, M.; Madden, V.J.; Welsch, C.; Antes, I.; Wen, Y.; Chugh, P.E.; McGee, C.E.; et al. Regulation of the Hepatitis C Virus RNA Replicase by Endogenous Lipid Peroxidation. Nat. Med. 2014, 20, 927–935. [Google Scholar] [CrossRef] [Green Version]
- Park, C.-Y.; Jun, H.-J.; Wakita, T.; Cheong, J.H.; Hwang, S.B. Hepatitis C Virus Nonstructural 4B Protein Modulates Sterol Regulatory Element-Binding Protein Signaling via the AKT Pathway. J. Biol. Chem. 2009, 284, 9237–9246. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Hood, B.L.; Chadwick, S.L.; Liu, S.; Watkins, S.C.; Luo, G.; Conrads, T.P.; Wang, T. Fatty Acid Synthase Is Up-Regulated during Hepatitis C Virus Infection and Regulates Hepatitis C Virus Entry and Production. Hepatology 2008, 48, 1396–1403. [Google Scholar] [CrossRef] [Green Version]
- Nasheri, N.; Joyce, M.; Rouleau, Y.; Yang, P.; Yao, S.; Tyrrell, D.L.; Pezacki, J.P. Modulation of Fatty Acid Synthase Enzyme Activity and Expression during Hepatitis C Virus Replication. Chem. Biol. 2013, 20, 570–582. [Google Scholar] [CrossRef]
- Borawski, J.; Troke, P.; Puyang, X.; Gibaja, V.; Zhao, S.; Mickanin, C.; Leighton-Davies, J.; Wilson, C.J.; Myer, V.; Cornellataracido, I.; et al. Class III Phosphatidylinositol 4-Kinase Alpha and Beta Are Novel Host Factor Regulators of Hepatitis C Virus Replication. J. Virol. 2009, 83, 10058–10074. [Google Scholar] [CrossRef] [Green Version]
- Hsu, N.-Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.-H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral Reorganization of the Secretory Pathway Generates Distinct Organelles for RNA Replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, K.L.; Kelly, S.M.; Jordan, T.X.; Tartell, M.A.; Randall, G. Hepatitis C Virus Stimulates the Phosphatidylinositol 4-Kinase III Alpha-Dependent Phosphatidylinositol 4-Phosphate Production That Is Essential for Its Replication. J. Virol. 2011, 85, 8870–8883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Hong, Z.; Lin, W.; Shao, R.-X.; Goto, K.; Hsu, V.W.; Chung, R.T. ARF1 and GBF1 Generate a PI4P-Enriched Environment Supportive of Hepatitis C Virus Replication. PLoS ONE 2012, 7, e32135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furse, S.; Brooks, N.J.; Seddon, A.M.; Woscholski, R.; Templer, R.H.; Tate, E.W.; Gaffney, P.R.J.; Ces, O. Lipid Membrane Curvature Induced by Distearoyl Phosphatidylinositol 4-Phosphate. Soft Matter 2012, 8, 3090–3093. [Google Scholar] [CrossRef]
- Godi, A.; Pertile, P.; Meyers, R.; Marra, P.; Di Tullio, G.; Iurisci, C.; Luini, A.; Corda, D.; De Matteis, M.A. ARF Mediates Recruitment of PtdIns-4-OH Kinase-Beta and Stimulates Synthesis of PtdIns(4,5)P2 on the Golgi Complex. Nat. Cell Biol. 1999, 1, 280–287. [Google Scholar] [CrossRef]
- Bishé, B.; Syed, G.; Siddiqui, A. Phosphoinositides in the Hepatitis C Virus Life Cycle. Viruses 2012, 4, 2340–2358. [Google Scholar] [CrossRef] [Green Version]
- Blumental-Perry, A.; Haney, C.J.; Weixel, K.M.; Watkins, S.C.; Weisz, O.A.; Aridor, M. Phosphatidylinositol 4-Phosphate Formation at ER Exit Sites Regulates ER Export. Dev. Cell 2006, 11, 671–682. [Google Scholar] [CrossRef]
- Bianco, A.; Reghellin, V.; Donnici, L.; Fenu, S.; Alvarez, R.; Baruffa, C.; Peri, F.; Pagani, M.; Abrignani, S.; Neddermann, P.; et al. Metabolism of Phosphatidylinositol 4-Kinase IIIα-Dependent PI4P Is Subverted by HCV and Is Targeted by a 4-Anilino Quinazoline with Antiviral Activity. PLoS Pathog. 2012, 8, e1002576. [Google Scholar] [CrossRef]
- Lim, Y.-S.; Hwang, S.B. Hepatitis C Virus NS5A Protein Interacts with Phosphatidylinositol 4-Kinase Type IIIalpha and Regulates Viral Propagation. J. Biol. Chem. 2011, 286, 11290–11298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Chung, K.-S.; Kim, D.-U.; Won, M.; Kim, L.; Kim, K.-S.; Nam, M.; Choi, S.-J.; Kim, H.-C.; Yoon, M.; et al. Systematic Identification of Hepatocellular Proteins Interacting with NS5A of the Hepatitis C Virus. J. Biochem. Mol. Biol. 2004, 37, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Reiss, S.; Harak, C.; Romero-Brey, I.; Radujkovic, D.; Klein, R.; Ruggieri, A.; Rebhan, I.; Bartenschlager, R.; Lohmann, V. The Lipid Kinase Phosphatidylinositol-4 Kinase III Alpha Regulates the Phosphorylation Status of Hepatitis C Virus NS5A. PLoS Pathog. 2013, 9, e1003359. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Marcotrigiano, J.; Rice, C.M. Structure of the Zinc-Binding Domain of an Essential Component of the Hepatitis C Virus Replicase. Nature 2005, 435, 374–379. [Google Scholar] [CrossRef] [Green Version]
- Love, R.A.; Brodsky, O.; Hickey, M.J.; Wells, P.A.; Cronin, C.N. Crystal Structure of a Novel Dimeric Form of NS5A Domain I Protein from Hepatitis C Virus. J. Virol. 2009, 83, 4395–4403. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.M.; Langley, D.R.; Garnett, J.A.; Angell, R.; Hedgethorne, K.; Meanwell, N.A.; Matthews, S.J. The Crystal Structure of NS5A Domain 1 from Genotype 1a Reveals New Clues to the Mechanism of Action for Dimeric HCV Inhibitors. Protein Sci. Publ. Protein Soc. 2014, 23, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Harak, C.; Radujkovic, D.; Taveneau, C.; Reiss, S.; Klein, R.; Bressanelli, S.; Lohmann, V. Mapping of Functional Domains of the Lipid Kinase Phosphatidylinositol 4-Kinase Type III Alpha Involved in Enzymatic Activity and Hepatitis C Virus Replication. J. Virol. 2014, 88, 9909–9926. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.-T.; Chen, S.S. Hepatitis C Virus Subverts Human Choline Kinase-α To Bridge Phosphatidylinositol-4-Kinase IIIα (PI4KIIIα) and NS5A and Upregulates PI4KIIIα Activation, Thereby Promoting the Translocation of the Ternary Complex to the Endoplasmic Reticulum for Viral Replication. J. Virol. 2017, 91, e00355-17. [Google Scholar] [CrossRef] [Green Version]
- Reghellin, V.; Donnici, L.; Fenu, S.; Berno, V.; Calabrese, V.; Pagani, M.; Abrignani, S.; Peri, F.; De Francesco, R.; Neddermann, P. NS5A Inhibitors Impair NS5A-Phosphatidylinositol 4-Kinase IIIα Complex Formation and Cause a Decrease of Phosphatidylinositol 4-Phosphate and Cholesterol Levels in Hepatitis C Virus-Associated Membranes. Antimicrob. Agents Chemother. 2014, 58, 7128–7140. [Google Scholar] [CrossRef] [Green Version]
- Altan-Bonnet, N.; Balla, T. Phosphatidylinositol 4-Kinases: Hostages Harnessed to Build Panviral Replication Platforms. Trends Biochem. Sci. 2012, 37, 293–302. [Google Scholar] [CrossRef]
- Li, H.; Yang, X.; Yang, G.; Hong, Z.; Zhou, L.; Yin, P.; Xiao, Y.; Chen, L.; Chung, R.T.; Zhang, L. Hepatitis C Virus NS5A Hijacks ARFGAP1 to Maintain a Phosphatidylinositol 4-Phosphate-Enriched Microenvironment. J. Virol. 2014, 88, 5956–5966. [Google Scholar] [CrossRef] [Green Version]
- Siu, G.K.Y.; Zhou, F.; Yu, M.K.; Zhang, L.; Wang, T.; Liang, Y.; Chen, Y.; Chan, H.C.; Yu, S. Hepatitis C Virus NS5A Protein Cooperates with Phosphatidylinositol 4-Kinase IIIα to Induce Mitochondrial Fragmentation. Sci. Rep. 2016, 6, 23464. [Google Scholar] [CrossRef] [Green Version]
- Brass, V.; Bieck, E.; Montserret, R.; Wölk, B.; Hellings, J.A.; Blum, H.E.; Penin, F.; Moradpour, D. An Amino-Terminal Amphipathic Alpha-Helix Mediates Membrane Association of the Hepatitis C Virus Nonstructural Protein 5A. J. Biol. Chem. 2002, 277, 8130–8139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penin, F.; Brass, V.; Appel, N.; Ramboarina, S.; Montserret, R.; Ficheux, D.; Blum, H.E.; Bartenschlager, R.; Moradpour, D. Structure and Function of the Membrane Anchor Domain of Hepatitis C Virus Nonstructural Protein 5A. J. Biol. Chem. 2004, 279, 40835–40843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Jackman, J.A.; Cho, N.-J. Comparing the Membrane-Interaction Profiles of Two Antiviral Peptides: Insights into Structure–Function Relationship. Langmuir 2019, 35, 9934–9943. [Google Scholar] [CrossRef] [PubMed]
- Feuerstein, S.; Solyom, Z.; Aladağ, A.; Hoffmann, S.; Willbold, D.; Brutscher, B. 1H, 13C, and 15N Resonance Assignment of a 179 Residue Fragment of Hepatitis C Virus Non-Structural Protein 5A. Biomol. NMR Assign. 2011, 5, 241–243. [Google Scholar] [CrossRef]
- Feuerstein, S.; Solyom, Z.; Aladag, A.; Favier, A.; Schwarten, M.; Hoffmann, S.; Willbold, D.; Brutscher, B. Transient Structure and SH3 Interaction Sites in an Intrinsically Disordered Fragment of the Hepatitis C Virus Protein NS5A. J. Mol. Biol. 2012, 420, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Jirasko, V.; Lends, A.; Lakomek, N.-A.; Fogeron, M.-L.; Weber, M.; Malär, A.; Penzel, S.; Bartenschlager, R.; Meier, B.H.; Böckmann, A. Dimer Organization of Membrane-Associated NS5A of Hepatitis C Virus as Determined by Highly Sensitive 1H-Detected Solid-State NMR. Angew. Chem. Int. Ed. Engl. 2021, 133, 5399–5407. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins in Human Diseases: Introducing the D2 Concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Cordek, D.G.; Bechtel, J.T.; Maynard, A.T.; Kazmierski, W.M.; Cameron, C.E. Targeting the ns5a protein of hcv: An emerging option. Drugs Future 2011, 36, 691–711. [Google Scholar] [CrossRef]
- Dujardin, M.; Madan, V.; Montserret, R.; Ahuja, P.; Huvent, I.; Launay, H.; Leroy, A.; Bartenschlager, R.; Penin, F.; Lippens, G.; et al. A Proline-Tryptophan Turn in the Intrinsically Disordered Domain 2 of NS5A Protein Is Essential for Hepatitis C Virus RNA Replication. J. Biol. Chem. 2015, 290, 19104–19120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Ye, H.; Kang, C.B.; Yoon, H.S. Domain 2 of Nonstructural Protein 5A (NS5A) of Hepatitis C Virus Is Natively Unfolded. Biochemistry 2007, 46, 11550–11558. [Google Scholar] [CrossRef]
- Hanoulle, X.; Badillo, A.; Verdegem, D.; Penin, F.; Lippens, G. The Domain 2 of the HCV NS5A Protein Is Intrinsically Unstructured. Protein Pept. Lett. 2010, 17, 1012–1018. [Google Scholar] [CrossRef]
- Badillo, A.; Receveur-Brechot, V.; Sarrazin, S.; Cantrelle, F.-X.; Delolme, F.; Fogeron, M.-L.; Molle, J.; Montserret, R.; Bockmann, A.; Bartenschlager, R.; et al. Overall Structural Model of NS5A Protein from Hepatitis C Virus and Modulation by Mutations Confering Resistance of Virus Replication to Cyclosporin A. Biochemistry 2017, 56, 3029–3048. [Google Scholar] [CrossRef]
- Hanoulle, X.; Verdegem, D.; Badillo, A.; Wieruszeski, J.-M.; Penin, F.; Lippens, G. Domain 3 of Non-Structural Protein 5A from Hepatitis C Virus Is Natively Unfolded. Biochem. Biophys. Res. Commun. 2009, 381, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Verdegem, D.; Badillo, A.; Wieruszeski, J.-M.; Landrieu, I.; Leroy, A.; Bartenschlager, R.; Penin, F.; Lippens, G.; Hanoulle, X. Domain 3 of NS5A Protein from the Hepatitis C Virus Has Intrinsic Alpha-Helical Propensity and Is a Substrate of Cyclophilin A. J. Biol. Chem. 2011, 286, 20441–20454. [Google Scholar] [CrossRef] [Green Version]
- Sólyom, Z.; Ma, P.; Schwarten, M.; Bosco, M.; Polidori, A.; Durand, G.; Willbold, D.; Brutscher, B. The Disordered Region of the HCV Protein NS5A: Conformational Dynamics, SH3 Binding, and Phosphorylation. Biophys. J. 2015, 109, 1483–1496. [Google Scholar] [CrossRef] [Green Version]
- Street, A.; Macdonald, A.; Crowder, K.; Harris, M. The Hepatitis C Virus NS5A Protein Activates a Phosphoinositide 3-Kinase-Dependent Survival Signaling Cascade. J. Biol. Chem. 2004, 279, 12232–12241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zech, B.; Kurtenbach, A.; Krieger, N.; Strand, D.; Blencke, S.; Morbitzer, M.; Salassidis, K.; Cotten, M.; Wissing, J.; Obert, S.; et al. Identification and Characterization of Amphiphysin II as a Novel Cellular Interaction Partner of the Hepatitis C Virus NS5A Protein. J. Gen. Virol. 2003, 84, 555–560. [Google Scholar] [CrossRef]
- Masumi, A.; Aizaki, H.; Suzuki, T.; DuHadaway, J.B.; Prendergast, G.C.; Komuro, K.; Fukazawa, H. Reduction of Hepatitis C Virus NS5A Phosphorylation through Its Interaction with Amphiphysin II. Biochem. Biophys. Res. Commun. 2005, 336, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Targett-Adams, P.; Graham, E.J.S.; Middleton, J.; Palmer, A.; Shaw, S.M.; Lavender, H.; Brain, P.; Tran, T.D.; Jones, L.H.; Wakenhut, F.; et al. Small Molecules Targeting Hepatitis C Virus-Encoded NS5A Cause Subcellular Redistribution of Their Target: Insights into Compound Modes of Action. J. Virol. 2011, 85, 6353–6368. [Google Scholar] [CrossRef] [Green Version]
- Conte, I.; Giuliano, C.; Ercolani, C.; Narjes, F.; Koch, U.; Rowley, M.; Altamura, S.; De Francesco, R.; Neddermann, P.; Migliaccio, G.; et al. Synthesis and SAR of Piperazinyl-N-Phenylbenzamides as Inhibitors of Hepatitis C Virus RNA Replication in Cell Culture. Bioorgan. Med. Chem. Lett. 2009, 19, 1779–1783. [Google Scholar] [CrossRef]
- Lemm, J.A.; O’Boyle, D.; Liu, M.; Nower, P.T.; Colonno, R.; Deshpande, M.S.; Snyder, L.B.; Martin, S.W.; St Laurent, D.R.; Serrano-Wu, M.H.; et al. Identification of Hepatitis C Virus NS5A Inhibitors. J. Virol. 2010, 84, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Nettles, J.H.; Stanton, R.A.; Broyde, J.; Amblard, F.; Zhang, H.; Zhou, L.; Shi, J.; McBrayer, T.R.; Whitaker, T.; Coats, S.J.; et al. Asymmetric Binding to NS5A by Daclatasvir (BMS-790052) and Analogs Suggests Two Novel Modes of HCV Inhibition. J. Med. Chem. 2014, 57, 10031–10043. [Google Scholar] [CrossRef] [Green Version]
- McMahon, H.T.; Boucrot, E. Membrane Curvature at a Glance. J. Cell Sci. 2015, 128, 1065–1070. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bulankina, A.V.; Richter, R.M.; Welsch, C. Regulatory Role of Phospholipids in Hepatitis C Virus Replication and Protein Function. Pathogens 2022, 11, 102. https://doi.org/10.3390/pathogens11010102
Bulankina AV, Richter RM, Welsch C. Regulatory Role of Phospholipids in Hepatitis C Virus Replication and Protein Function. Pathogens. 2022; 11(1):102. https://doi.org/10.3390/pathogens11010102
Chicago/Turabian StyleBulankina, Anna V., Rebecca M. Richter, and Christoph Welsch. 2022. "Regulatory Role of Phospholipids in Hepatitis C Virus Replication and Protein Function" Pathogens 11, no. 1: 102. https://doi.org/10.3390/pathogens11010102