
The Population Diversity of Candidate Genes for Resistance/Susceptibility to Coronavirus Infection in Domestic Cats: An Inter-Breed Comparison


 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Within-Breed Diversity of Candidate Genes
2.2. Inter-Breed Comparisons
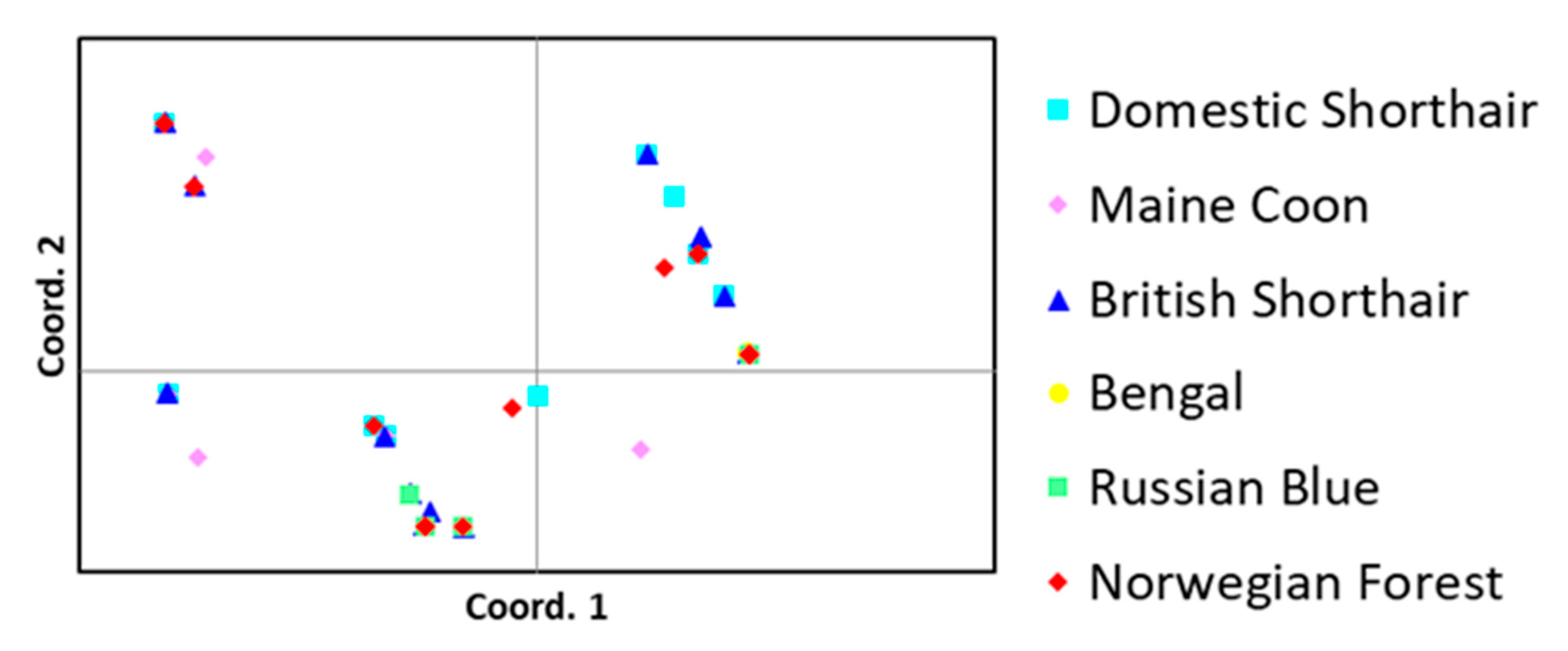
2.3. FST-Based Comparisons, PCoA and STRUCTURE Analyses
2.3.1. FST-Based Comparisons
2.3.2. PCoA and STRUCTURE Analyses
2.4. Haplotypes
3. Discussion
4. Materials and Methods
4.1. Cats
4.2. Candidate Genes
4.3. Genotyping
4.4. Data Analysis and SNP Calling
4.5. Parameters of Population Diversity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kennedy, M.A. Feline Infectious Peritonitis. Veter. Clin. N. Am. Small Anim. Pract. 2020, 50, 1001–1011. [Google Scholar] [CrossRef]
- Addie, D.D.; Toth, S.; Murray, G.D.; Jarrett, O. Risk of feline infectious peritonitis in cats naturally infected with feline coro-navirus. Am. J. Vet. Res. 1995, 56, 429–434. [Google Scholar] [PubMed]
- Kipar, A.; Meli, M.L. Feline Infectious Peritonitis. Veter. Pathol. 2014, 51, 505–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, E.; Tasker, S.; Day, M.J.; Harley, R.; Kipar, A.; Siddell, S.G.; Helps, C.R. Amino acid changes in the spike protein of feline coronavirus correlate with systemic spread of virus from the intestine and not with feline infectious peritonitis. Veter. Res. 2014, 45, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaimes, J.A.; Millet, J.K.; Stout, A.E.; André, N.M.; Whittaker, G.R. A Tale of Two Viruses: The Distinct Spike Glycoproteins of Feline Coronaviruses. Viruses 2020, 12, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malbon, A.J.; Russo, G.; Burgener, C.; Barker, E.N.; Meli, M.L.; Tasker, S.; Kipar, A. The Effect of Natural Feline Coronavirus Infection on the Host Immune Response: A Whole-Transcriptome Analysis of the Mesenteric Lymph Nodes in Cats with and without Feline Infectious Peritonitis. Pathogens 2020, 9, 524. [Google Scholar] [CrossRef]
- Pedersen, N.C.; Allen, C.E.; Lyons, L.A. Pathogenesis of feline enteric coronavirus infection. J. Feline Med. Surg. 2008, 10, 529–541. [Google Scholar] [CrossRef] [Green Version]
- Addie, D.D.; Jarrett, O. A study of naturally occurring feline coronavirus infections in kittens. Veter. Rec. 1992, 130, 133–137. [Google Scholar] [CrossRef]
- LoPresti, M.; Beck, D.B.; Duggal, P.; Cummings, D.A.; Solomon, B.D. The Role of Host Genetic Factors in Coronavirus Susceptibility: Review of Animal and Systematic Review of Human Literature. Am. J. Hum. Genet. 2020, 107, 381–402. [Google Scholar] [CrossRef] [PubMed]
- Golovko, L.; Lyons, L.A.; Liu, H.; Sørensen, A.; Wehnert, S.; Pedersen, N.C. Genetic susceptibility to feline infectious peritonitis in Birman cats. Virus Res. 2013, 175, 58–63. [Google Scholar] [CrossRef]
- Pedersen, N.C.; Liu, H.; Durden, M.; Lyons, L.A. Natural resistance to experimental feline infectious peritonitis virus infection is decreased rather than increased by positive genetic selection. Veter. Immunol. Immunopathol. 2016, 171, 17–20. [Google Scholar] [CrossRef] [Green Version]
- Kiss, I.; Kecskeméti, S.; Tanyi, J.; Klingeborn, B.; Belák, S. Prevalence and Genetic Pattern of Feline Coronaviruses in Urban Cat Populations. Veter. J. 2000, 159, 64–70. [Google Scholar] [CrossRef]
- Norris, J.M.; Bosward, K.L.; White, J.D.; Baral, R.M.; Catt, M.J.; Malik, R. Clinicopathological findings associated with feline infectious peritonitis in Sydney, Australia: 42 cases (1990–2002). Aust. Veter. J. 2005, 83, 666–673. [Google Scholar] [CrossRef]
- Barker, E.N.; Lait, P.; Ressel, L.; Blackwell, E.-J.; Tasker, S.; Kedward-Dixon, H.; Kipar, A.; Helps, C.R. Evaluation of Interferon-Gamma Polymorphisms as a Risk Factor in Feline Infectious Peritonitis Development in Non-Pedigree Cats—A Large Cohort Study. Pathogens 2020, 9, 535. [Google Scholar] [CrossRef]
- Kedward-Dixon, H.; Barker, E.N.; Tasker, S.; Kipar, A.; Helps, C.R. Evaluation of polymorphisms in inflammatory mediator and cellular adhesion genes as risk factors for feline infectious peritonitis. J. Feline Med. Surg. 2019, 22, 564–570. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-T.; Hsieh, L.-E.; Dai, Y.-R.; Chueh, L.-L. Polymorphisms in the feline TNFA and CD209 genes are associated with the outcome of feline coronavirus infection. Veter. Res. 2014, 45, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, N.C.; Liu, H.; Gandolfi, B.; Lyons, L.A. The influence of age and genetics on natural resistance to experimentally induced feline infectious peritonitis. Veter. Immunol. Immunopathol. 2014, 162, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Addie, D.D.; Kennedy, L.J.; Ryvar, R.; Willoughby, K.; Gaskell, R.M.; Ollier, W.; Nart, P.; Radford, A. Feline leucocyte antigen class II polymorphism and susceptibility to feline infectious peritonitis. J. Feline Med. Surg. 2004, 6, 59–62. [Google Scholar] [CrossRef]
- Bubenikova, J.; Vrabelova, J.; Stejskalova, K.; Futas, J.; Plasil, M.; Cerna, P.; Oppelt, J.; Lobova, D.; Molinkova, D.; Horin, P. Candidate Gene Markers Associated with Fecal Shedding of the Feline Enteric Coronavirus (FECV). Pathogens 2020, 9, 958. [Google Scholar] [CrossRef]
- Vermeulen, B.L.; Devriendt, B.; Olyslaegers, D.A.; Dedeurwaerder, A.; Desmarets, L.M.; Favoreel, H.W.; Dewerchin, H.L.; Nauwynck, H.J. Suppression of NK cells and regulatory T lymphocytes in cats naturally infected with feline infectious peritonitis virus. Veter. Microbiol. 2013, 164, 46–59. [Google Scholar] [CrossRef]
- Pedersen, N.C. A review of feline infectious peritonitis virus infection: 1963–2008. J. Feline Med. Surg. 2009, 11, 225–258. [Google Scholar] [CrossRef] [PubMed]
- Addie, D.; Belák, S.; Boucraut-Baralon, C.; Egberink, H.; Frymus, T.; Gruffydd-Jones, T.; Hartmann, K.; Hosie, M.J.; Lloret, A.; Lutz, H.; et al. Feline infectious peritonitis. ABCD guidelines on prevention and management. J. Feline Med. Surg. 2009, 11, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Drechsler, Y.; Alcaraz, A.; Bossong, F.J.; Collisson, E.W.; Diniz, P.P.V. Feline Coronavirus in Multicat Environments. Veter. Clin. N. Am. Small Anim. Pract. 2011, 41, 1133–1169. [Google Scholar] [CrossRef] [PubMed]
- Nezi, M.; Mastorakos, G.; Mouslech, Z. Corticotropin Releasing Hormone And The Immune/Inflammatory Response. [Up-dated 2015 Jul 30]. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK279017/ (accessed on 12 March 2021).
- Worthing, K.A.; Wigney, D.I.; Dhand, N.K.; Fawcett, A.; McDonagh, P.; Malik, R.; Norris, J.M. Risk factors for feline infectious peritonitis in Australian cats. J. Feline Med. Surg. 2012, 14, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Pesteanu-Somogyi, L.D.; Radzai, C.; Pressler, B.M. Prevalence of feline infectious peritonitis in specific cat breeds. J. Feline Med. Surg. 2006, 8, 1–5. [Google Scholar] [CrossRef]
- Soma, T.; Wada, M.; Taharaguchi, S.; Tajima, T. Detection of Ascitic Feline Coronavirus RNA from Cats with Clinically Suspected Feline Infectious Peritonitis. J. Veter. Med. Sci. 2013, 75, 1389–1392. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Salazar, C.; Sun, J.C. Natural killer cell responses to emerging viruses of zoonotic origin. Curr. Opin. Virol. 2020, 44, 97–111. [Google Scholar] [CrossRef]
- Watanabe, R.; Eckstrand, C.; Liu, H.; Pedersen, N.C. Characterization of peritoneal cells from cats with experimentally-induced feline infectious peritonitis (FIP) using RNA-seq. Veter. Res. 2018, 49, 1–15. [Google Scholar] [CrossRef] [Green Version]
- FastQC [Computer Software]. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 8 March 2016).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Breese, M.; Liu, Y. NGSUtils: A software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics 2013, 29, 494–496. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Picard Tools [Computer Software]. Available online: https://broadinstitute.github.io/picard (accessed on 6 February 2016).
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Bam-Readcount [Computer Software]. Available online: https://github.com/genome/bam-readcount (accessed on 21 October 2016).
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2011, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | CRH | CRHR1 | CRHBP | PRF1 | GZMA | GZMB | IFNL | IFNLR |
|---|---|---|---|---|---|---|---|---|
| Numbers of interbreed differences | 11 | 12 | 12 | 11 | 13 | 14 | 9 | 13 |
| Gene | NCR1 | NCR2 | NCR3 | LAMP5 | SNX5 | SLX4IP | PAK5 | PCSK |
| Numbers of interbreed differences | 14 | 14 | 6 | 8 | 8 | 10 | 7 | 14 |
| Domestic Shorthair | Maine Coon | British Shorthair | Bengal | Russian Blue | Norwegian Forest | |
|---|---|---|---|---|---|---|
| 0 | 0.153 | 0.106 | 0.142 | 0.247 | 0.037 | Domestic Shorthair |
| 0.057 | 0 | 0.193 | 0.240 | 0.215 | 0.199 | Maine Coon |
| 0.090 | 0.098 | 0 | 0.286 | 0.180 | 0.039 | British Shorthair |
| 0.190 | 0.236 | 0.225 | 0 | 0.413 | 0.249 | Bengal |
| 0.199 | 0.230 | 0.209 | 0.355 | 0 | 0.232 | Russian Blue |
| 0.116 | 0.161 | 0.101 | 0.190 | 0.225 | 0 | Norwegian Forest |
| Allele/Genotype Frequency | All SNPs | Associated SNPs | ||||||
|---|---|---|---|---|---|---|---|---|
| Gene | Total | Cluster 1 vs. 2 | Cluster 1 vs. 3 | Cluster 2 vs. 3 | Total | Cluster 1 vs. 2 | Cluster 1 vs. 3 | Cluster 2 vs. 3 |
| NCR1 (p < 0.01) | 38 | 13 | 17 | 11 | 1 | 1 | 1 | 1 |
| SLX4IP (p < 0.05) | 6 | 0 | 0 | 1 | 3 | 0 | 0 | 2 |
| Both genes altogether | 44 | 13 | 17 | 12 | 4 | 1 | 1 | 3 |
| Homozygosity | All SNPs | Associated SNPs | ||||||
| Gene | Total | Cluster 1 vs. 2 | Cluster 1 vs. 3 | Cluster 2 vs. 3 | Total | Cluster 1 vs. 2 | Cluster 1 vs. 3 | Cluster 2 vs. 3 |
| NCR1 (p < 0.01) | 38 | 6 | 0 | 10 | 1 | 0 | 1 | 1 |
| SLX4IP (p < 0.05) | 6 | 0 | 0 | 1 | 3 | 0 | 0 | 0 |
| Both genes altogether | 44 | 6 | 0 | 11 | 4 | 0 | 1 | 1 |
| Breeds | Bengal | British Shorthair | Domestic Shorthair | Maine Coon | Norwegian Forest | Russian Blue |
|---|---|---|---|---|---|---|
| Bengal | -- | 0 | 0 | 1 | 0 | 0 |
| British Shorthair | 1 | -- | 0 | 1 | 0 | 0 |
| Domestic Shorthair | 0 | 1 | -- | 0 | 0 | 0 |
| Maine Coon | 0 | 1 | 0 | -- | 1 | 1 |
| Norwegian Forest | 1 | 1 | 0 | 1 | -- | 0 |
| Russian Blue | 1 | 1 | 1 | 1 | 1 | -- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bubenikova, J.; Vychodilova, L.; Stejskalova, K.; Futas, J.; Oppelt, J.; Cerna, P.; Plasil, M.; Horin, P. The Population Diversity of Candidate Genes for Resistance/Susceptibility to Coronavirus Infection in Domestic Cats: An Inter-Breed Comparison. Pathogens 2021, 10, 778. https://doi.org/10.3390/pathogens10060778
Bubenikova J, Vychodilova L, Stejskalova K, Futas J, Oppelt J, Cerna P, Plasil M, Horin P. The Population Diversity of Candidate Genes for Resistance/Susceptibility to Coronavirus Infection in Domestic Cats: An Inter-Breed Comparison. Pathogens. 2021; 10(6):778. https://doi.org/10.3390/pathogens10060778
Chicago/Turabian StyleBubenikova, Jana, Leona Vychodilova, Karla Stejskalova, Jan Futas, Jan Oppelt, Petra Cerna, Martin Plasil, and Petr Horin. 2021. "The Population Diversity of Candidate Genes for Resistance/Susceptibility to Coronavirus Infection in Domestic Cats: An Inter-Breed Comparison" Pathogens 10, no. 6: 778. https://doi.org/10.3390/pathogens10060778