
Enhancing the Protective Immune Response to Administration of a LIVP-GFP Live Attenuated Vaccinia Virus to Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Creation of Recombinant Viruses
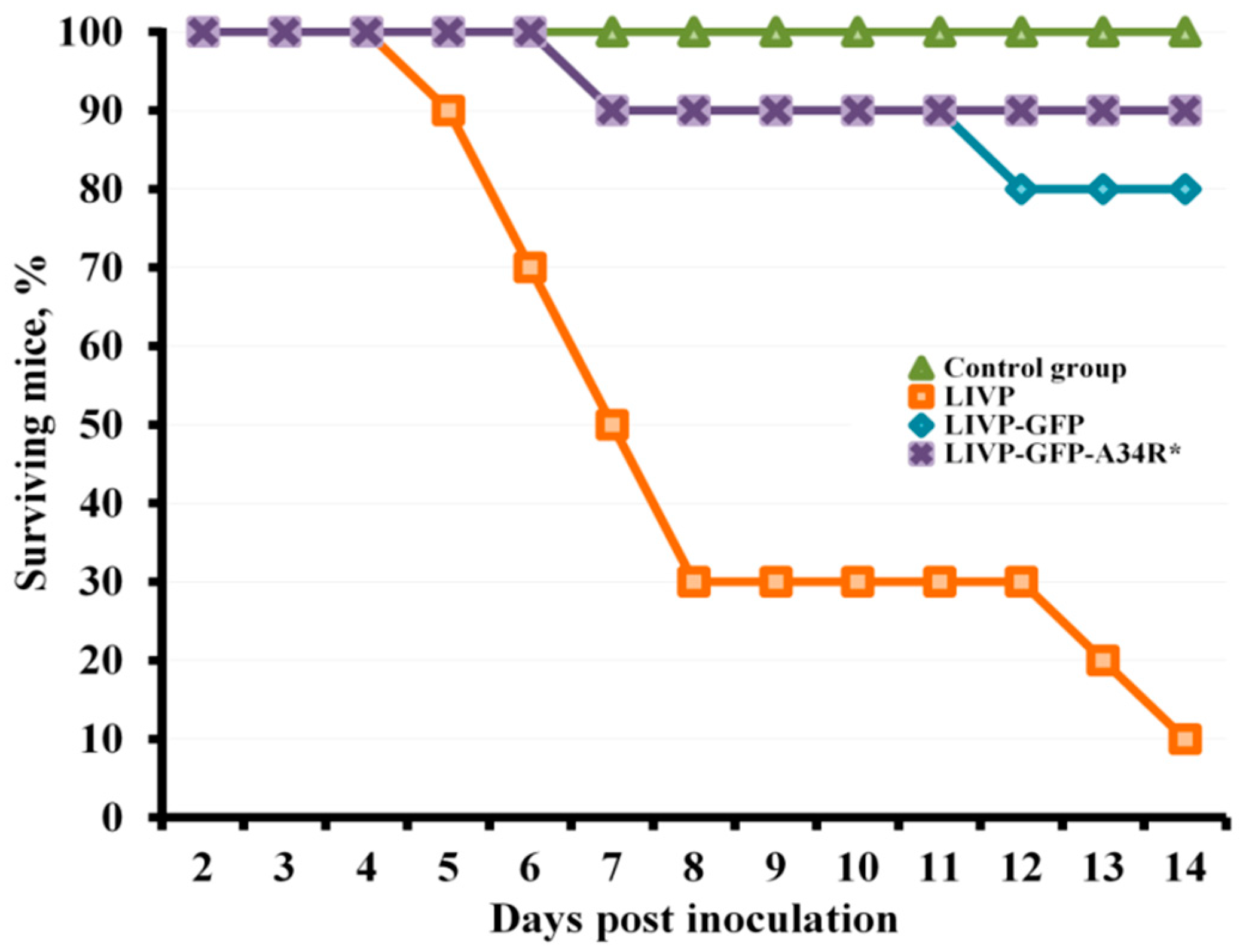
2.2. LIVP-GFP and LIVP-GFP-A34R* Viruses Exhibit Reduced Neurovirulence
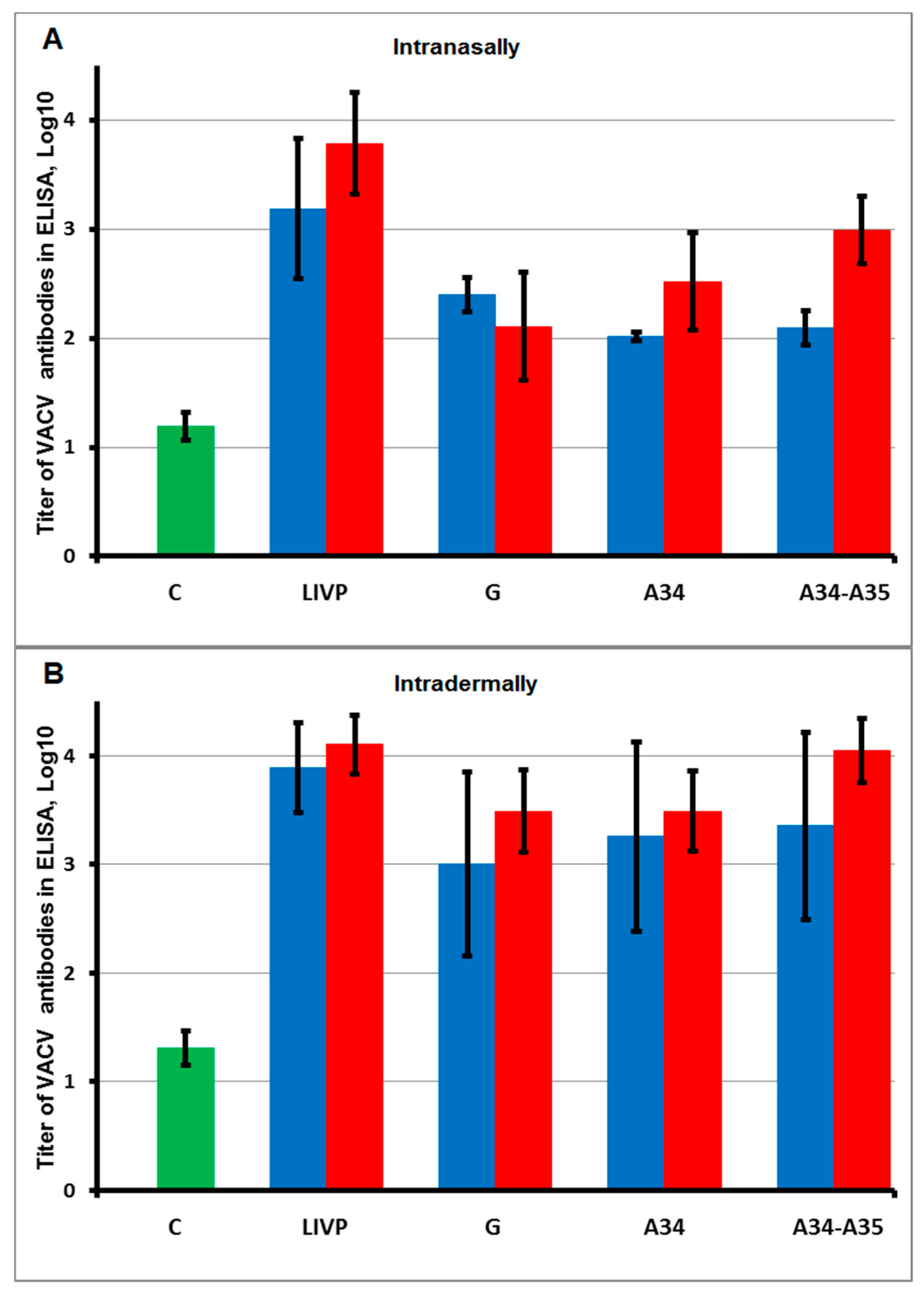
2.3. Intradermal Injection of VACV LIVP Variants Provides Higher Production of Virus-Specific Antibodies Compared to Intranasal Administration
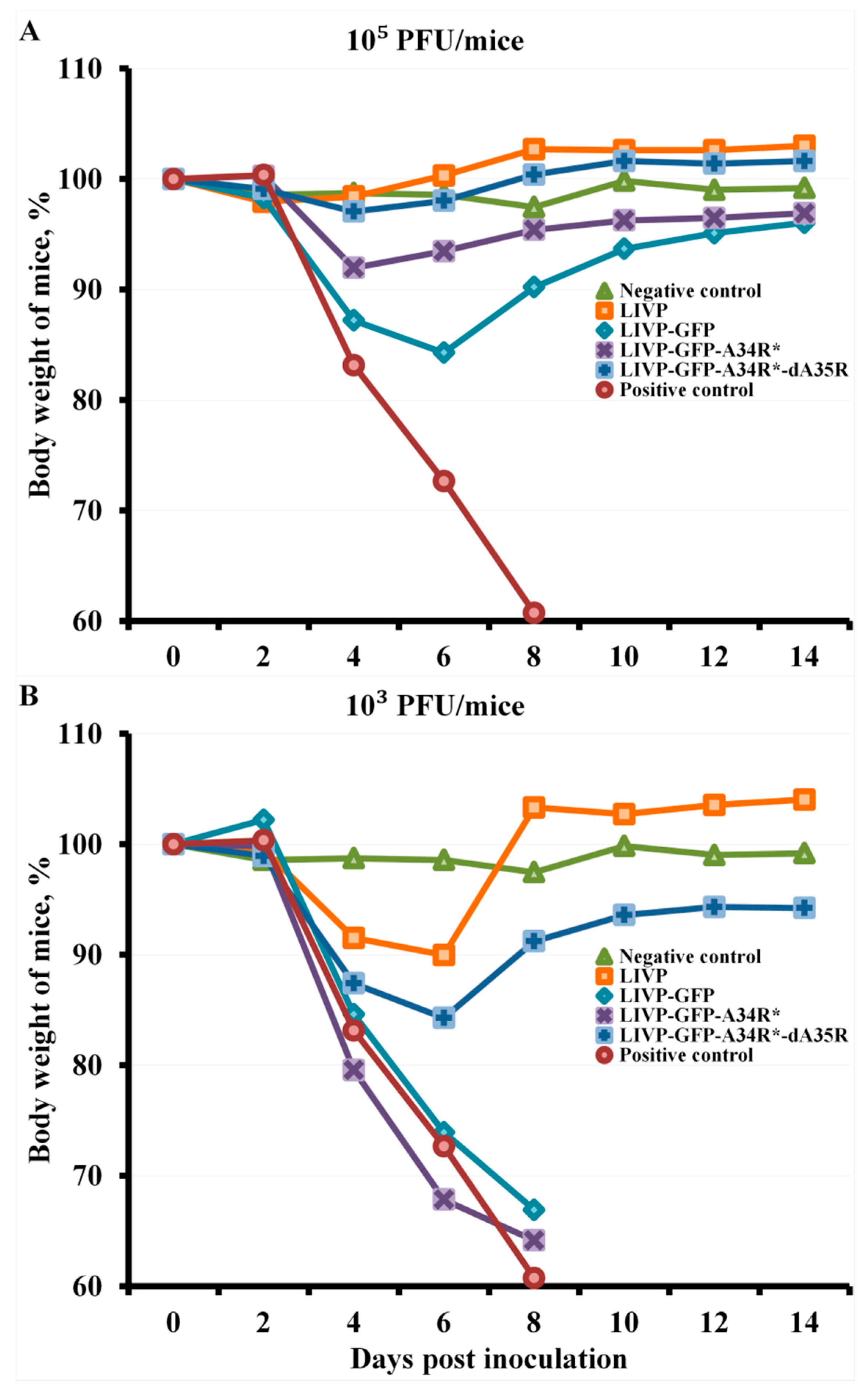
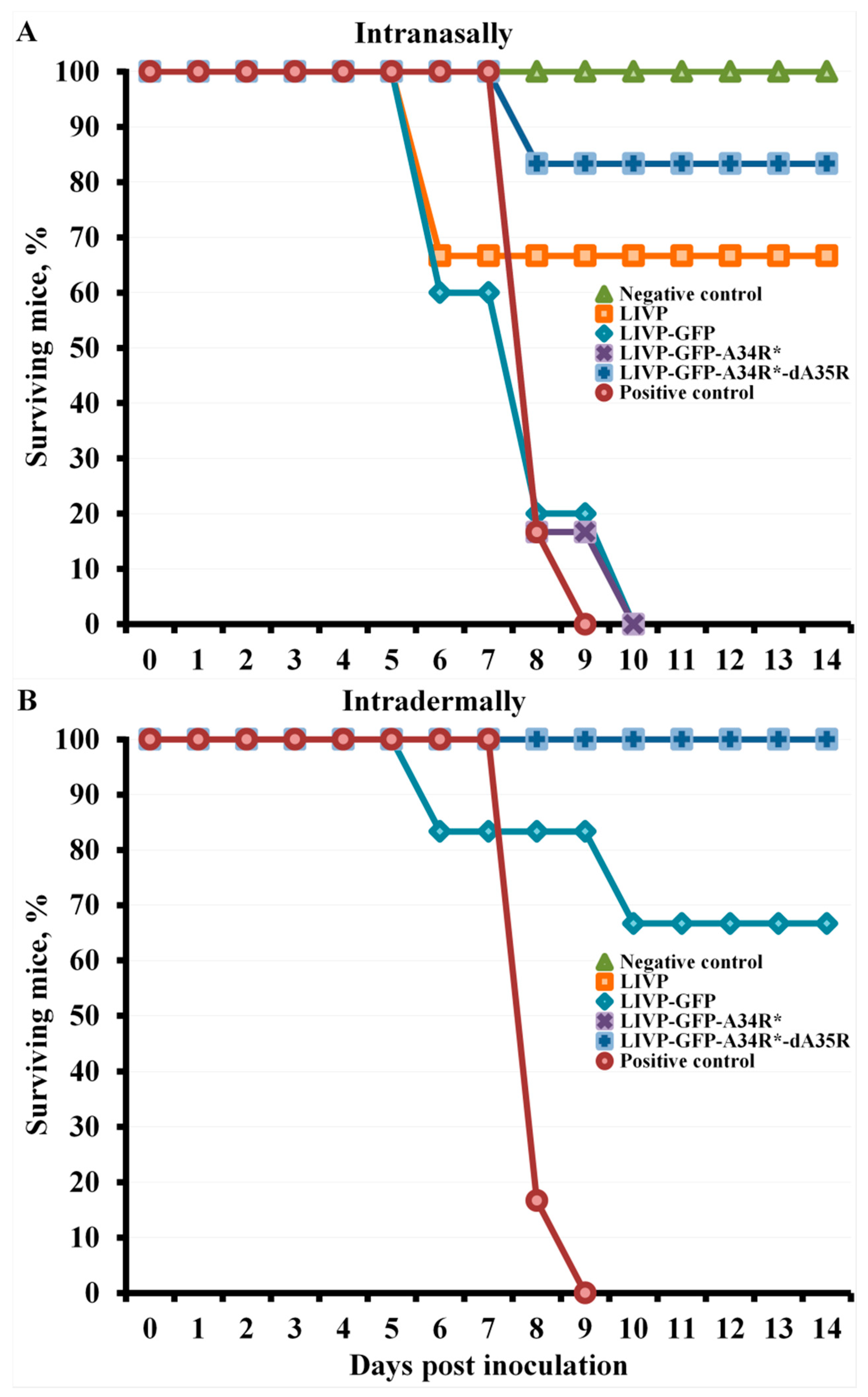
2.4. Protection against Orthopoxvirus Infection Depends on the Route of Administration and the Dose of Viruses
3. Discussion
4. Materials and Methods
4.1. Viruses
4.2. Animals
4.3. Sampling Mouse Serum
4.4. Assessment of the Protective Potency in Immunized Mice
4.5. Assessment of Viral Neurovirulence
4.6. Assessment of Lethality of Cowpox Virus
4.7. Enzyme-Linked Immunosorbent Assay of Mouse Serum
4.8. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shchelkunov, S.N.; Shchelkunova, G.A. Genes that control vaccinia virus immunogenicity. Acta Nat. 2020, 12, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Shchelkunov, S.N. An increasing danger of zoonotic orthopoxvirus infections. PLoS Pathog. 2013, 9, e1003756. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Sampedro, L.; Perdiguero, B.; Mejias-Perez, E.; Garcia-Arriaza, J.; Di Pilato, M.; Esteban, M. The evolution of poxvirus vaccines. Viruses 2015, 7, 1726–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shchelkunov, S.N.; Marennikova, S.S.; Moyer, R.W. Orthopoxviruses Pathogenic for Humans; Springer: New York, NY, USA, 2005; pp. 65–87. [Google Scholar]
- Kretzschmar, M.; Wallinga, J.; Teunis, P.; Xing, S.; Mikolajczyk, R. Frequency of adverse events after vaccination with different vaccinia strains. PLoS Med. 2006, 3, e272. [Google Scholar] [CrossRef]
- Jacobs, B.L.; Langland, J.O.; Kibler, K.V.; Denzler, K.L.; White, S.D.; Holechek, S.A.; Wong, S.; Huynh, T.; Baskin, C.R. Vaccinia virus vaccines: Past, present and future. Antiviral Res. 2009, 84, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Fenner, F.; Henderson, D.A.; Arita, I.; Jezek, Z.; Ladnyi, I.D. Smallpox and Its Eradication; World Health Organization: Geneva, Switzerland, 1988; 1460p. [Google Scholar]
- World Health Assembly. Global Smallpox Eradication; World Health Organization: Geneva, Switzerland, 1980. [Google Scholar]
- Olson, V.A.; Shchelkunov, S.N. Are we prepared in case of a possible smallpox-like disease emergence? Viruses 2017, 9, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albarnaz, J.D.; Torres, A.A.; Smith, G.L. Modulating vaccinia virus immunomodulators to improve immunological memory. Viruses 2018, 10, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, M.G.; Doty, J.B.; McCollum, A.M.; Olson, V.A.; Nakazawa, Y. Monkeypox re-emergence in Africa: A call to expand the concept and practice of One Health. Expert Rev. Anti Infect. Ther. 2019, 17, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Styczynski, A.; Burgado, J.; Walteros, D.; Usme-Ciro, J.; Laiton, K.; Farias, A.P.; Nakazawa, Y.; Chapman, C.; Davidson, W.; Mauldin, M.; et al. Seroprevalence and risk factors possibly associated with emerging zoonotic vaccinia virus in a farming community, Colombia. Emerg. Infect. Dis. 2019, 25, 2169–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, B. Smallpox vaccines: Targets of protective immunity. Immunol. Rev. 2011, 239, 8–26. [Google Scholar] [CrossRef]
- Shchelkunov, S.N. Emergence and reemergence of smallpox: The need in development of a new generation smallpox vaccine. Vaccine 2011, 29S, D49–D53. [Google Scholar] [CrossRef]
- Shchelkunov, S.N.; Nesterov, A.E.; Ryazankin, I.A.; Ignat’ev, G.M.; Sandakhchiev, L.S. Development of a candidate polyvalent live vaccine against human immunodeficiency, hepatitis B, and orthopoxviruses. Dokl. Biochem. Biophys. 2003, 390, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Shchelkunov, S.N.; Razumov, I.A.; Kolosova, I.V.; Romashchenko, A.V.; Zavjalov, E.L. Virotherapy of the malignant U87 human glioblastoma in the orthotopic xenotransplantation mouse SCID model. Dokl. Biochem. Biophys. 2018, 478, 30–33. [Google Scholar] [CrossRef]
- Li, J.; O’Malley, M.; Urban, J.; Sampath, P.; Guo, Z.S.; Kalinski, P.; Thorne, S.H.; Bartlett, D.L. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol. Ther. 2011, 19, 650–657. [Google Scholar] [CrossRef]
- Goncharova, E.P.; Ruzhenkova, J.S.; Petrov, I.S.; Shchelkunov, S.N.; Zenkova, M.A. Oncolytic virus efficiency inhibited growth of tumour cells with multiple drug resistant phenotype in vivo and in vitro. J. Transl. Med. 2016, 14, e241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhu, Y.; Chen, S.; Li, W.; Yin, X.; Li, S.; Xiao, P.; Han, J.; Li, X.; Sun, L.; et al. Generation of an attenuated Tiantan vaccinia virus strain by deletion of multiple genes. Front. Cell. Infect. Microbiol. 2017, 7, 462. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.S.; Lu, B.; Guo, Z.; Giehl, E.; Feist, M.; Dai, E.; Liu, W.; Storkus, W.J.; He, Y.; Liu, Z.; et al. Vaccinia virus-mediated cancer immunotherapy: Cancer vaccines and oncolytics. J. Immunother. Cancer 2019, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Rehm, K.E.; Connor, R.F.; Jones, G.J.B.; Yimbu, K.; Mannie, M.D.; Roper, R.L. Vaccinia virus decreases major histocompatibility complex (MHC) class II antigen presentation, T-cell priming, and peptide association with MHC class II. Immunology 2009, 128, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Rehm, K.E.; Connor, R.F.; Jones, G.J.B.; Yimbu, K.; Roper, R.L. Vaccinia virus A35R inhibits MHC class II antigen presentation. Virology 2010, 397, 176–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehm, K.E.; Roper, R.L. Deletion of the A35 gene from Modified Vaccinia Virus Ankara increases immunogenicity and isotype switching. Vaccine 2011, 29, 3276–3283. [Google Scholar] [CrossRef] [Green Version]
- Yakubitskiy, S.N.; Kolosova, I.V.; Maksyutov, R.A.; Shchelkunov, S.N. Highly immunogenic variant of attenuated vaccinia virus. Dokl. Biochem. Biophys. 2016, 466, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Vanderplasschen, A.; Law, M. The formation of extracellular enveloped vaccinia virus. J. Gen. Virol. 2002, 83, 2915–2931. [Google Scholar] [CrossRef]
- Payne, L.G. Significance of extracellular enveloped virus in the in vitro and in vivo dissemination of vaccinia. J. Gen. Virol. 1980, 50, 89–100. [Google Scholar] [CrossRef]
- Blasco, R.; Sisler, J.R.; Moss, B. Dissociation of progeny vaccinia virus from the cell membrane is regulated by a viral envelope glycoprotein: Effect of a point mutation in the lectin homology domain of the A34R gene. J. Virol. 1993, 67, 3319–3325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locker, J.K.; Kuehn, A.; Schleich, S.; Rutter, G.; Hohenberg, H.; Wepf, R.; Griffiths, G. Entry of the two infectious forms of vaccinia virus at the plasma membrane is signaling-dependent for IMV but not the EEV. Mol. Biol. Cell. 2000, 11, 2497–2511. [Google Scholar] [CrossRef] [Green Version]
- McNulty, S.; Powell, K.; Erneux, C.; Kalman, D. The host phosphoinositide 5-phosphatase SHIP2 regulates dissemination of vaccinia virus. J. Virol. 2011, 85, 7402–7410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monticelli, S.R.; Earley, A.K.; Tate, J.; Ward, B.M. The ectodomain of the vaccinia virus glycoprotein A34 is required for cell binding by extracellular virions and contains a large region capable of interaction with glycoprotein B5. J. Virol. 2019, 93, e01343-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.S.; Roos, J.M.; McGuigan, L.C.; Smith, K.A.; Cormier, N.; Cohen, L.K.; Roberts, B.E.; Payne, L.G. Molecular attenuation of vaccinia virus: Mutant generation and animal characterization. J. Virol. 1992, 66, 2617–2630. [Google Scholar] [CrossRef] [Green Version]
- Kirn, D.H.; Wang, Y.; Liang, W.; Contag, C.H.; Thorne, S.H. Enhancing poxvirus oncolytic effects through increased spread and immune evasion. Cancer Res. 2008, 68, 2071–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirunavukarasu, P.; Sathaiah, M.; Gorry, M.C.; O’Malley, M.E.; Ravindranathan, R.; Austin, F.; Thorne, S.H.; Guo, Z.S.; Bartlett, D.I. A rationally designed A34R mutant oncolytic poxvirus: Improved efficacy in peritoneal carcinomatosis. Mol. Ther. 2013, 21, 1024–1033. [Google Scholar] [CrossRef] [Green Version]
- Yakubitskiy, S.N.; Kolosova, I.V.; Maksyutov, R.A.; Shchelkunov, S.N. Attenuation of vaccinia virus. Acta Nat. 2015, 7, 113–121. [Google Scholar] [CrossRef]
- Petrov, I.S.; Goncharova, E.P.; Pozdnyakov, S.G.; Shchelkunov, S.N.; Zenkova, M.A.; Vlasov, V.V.; Kolosova, I.V. Antitumor effect of the LIVP-GFP recombinant vaccinia virus. Dokl. Biol. Sci. 2013, 45, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.V.; Tregubchak, T.V.; Shchelkunov, S.N.; Maksyutov, R.A.; Gavrilova, E.V. Obtaining vaccinia virus with increased production of extracellular enveloped virions and directing GM-CSF synthesis as a promising basis for development of antitumor drug. Med. Immunol. 2020, 22, 371–378. [Google Scholar] [CrossRef]
- Shchelkunov, S.N.; Safronov, P.F.; Totmenin, A.V.; Petrov, N.A.; Ryazankina, O.I.; Gutorov, V.V.; Kotwal, G.J. The genomic sequence analysis of the left and right species-specific terminal region of a cowpox virus strain reveals unique sequences and a cluster of intact ORFs for immunomodulatory and host range proteins. Virology 1998, 243, 432–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, M.D.; Kirchhoff, F.; Czajak, S.C.; Sehgal, P.K.; Desrosiers, R.C. Protective effects of a live attenuated SIV vaccine with a deletion in the nef gene. Science 1992, 258, 1938–1941. [Google Scholar] [CrossRef] [Green Version]
- Sanglard, D.; Hube, B.; Monod, M.; Odds, F.C.; Gow, N.A. A triple deletion of the secreted aspartyl proteinase genes SAP4, SAP5, and SAP6 of Candida albicans causes attenuated virulence. Infect. Immun. Actions 1997, 65, 3539–3546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambandamurthy, V.K.; Jacobs, W.R., Jr. Live attenuated mutants of Mycobacterium tuberculosis as candidate vaccines against tuberculosis. Microbes Infect. Actions 2005, 7, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Sattler, J.M.; Singer, M.; Heiss, K.; Reinig, M.; Hammerschmidt-Kamper, C.; Heussler, V.; Mueller, A.-K.; Frischknecht, F. Protective efficacy and safety of liver stage attenuated malaria parasites. Sci. Rep. Actions 2016, 6, 26824. [Google Scholar] [CrossRef] [Green Version]
- Breiman, A.; Carpentier, D.C.J.; Ewles, H.A.; Smith, G.L. Transport and stability of the vaccinia virus A34 protein is affected by the A33 protein. J. Gen. Virol. 2013, 94, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Earley, A.E.; Chan, W.M.; Ward, B.M. The vaccinia virus B5 protein requires A34 for efficient intracellular trafficking from the endoplasmic reticulum to the site of wrapping and incorporation into progeny virions. J. Virol. 2008, 82, 2161–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntosh, A.A.G.; Smith, G.L. Vaccinia virus glycoprotein A34R is required for infectivity of extracellular enveloped virus. J. Virol. 1996, 70, 272–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shchelkunov, S.N.; Yakubitskiy, S.N.; Bauer, T.V.; Sergeev, A.A.; Kabanov, A.S.; Bulichev, L.E.; Yurganova, I.A.; Odnoshevskiy, D.A.; Kolosova, I.V.; Pyankov, S.A.; et al. The influence of an elevated production of extracellular enveloped virions of the vaccinia virus on its properties in infected mice. Acta Nat. 2020, 12, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Strnadova, P.; Ren, H.; Valentine, R.; Mazzon, M.; Sweeney, T.R.; Brierley, I.; Smith, G.L. Inhibition of translation initiation by protein 169: A vaccinia virus strategy to suppress innate and adaptive immunity and alter virus virulence. PLoS Pathog. 2015, 11, e1005151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Rubin, S.A.; Taffs, R.E.; Merchlinsky, M.; Ye, Z.; Carbone, K.M. Mouse neurotoxicity test for vaccinia-based smallpox vaccines. Vaccine 2004, 22, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Shchelkunov, S.N.; Yakubitskiy, S.N.; Sergeev, A.A.; Kabanov, A.S.; Bauer, T.V.; Bulichev, L.E.; Pyankov, S.A. Effect of the route of administration of the vaccinia virus strain LIVP to mice on its virulence and immunogenicity. Viruses 2020, 12, 795. [Google Scholar] [CrossRef] [PubMed]
- Sachs, L. Statistische Auswertungsmethoden; Springer: Heidelberg, Germany, 1972; 193p. [Google Scholar]
- Mazurkov, O.Y.; Kabanov, A.S.; Shishkina, L.N.; Sergeev, A.A.; Skarnovich, M.O.; Bormotov, N.I.; Skarnovich, M.A.; Ovchinnikova, A.S.; Titova, K.A.; Galahova, D.O.; et al. New effective chemically synthesized anti-smallpox compound NIOCH-14. J. Gen. Virol. 2016, 97, 1229–1239. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shchelkunov, S.N.; Yakubitskiy, S.N.; Titova, K.A.; Pyankov, S.A.; Sergeev, A.A. Enhancing the Protective Immune Response to Administration of a LIVP-GFP Live Attenuated Vaccinia Virus to Mice. Pathogens 2021, 10, 377. https://doi.org/10.3390/pathogens10030377
Shchelkunov SN, Yakubitskiy SN, Titova KA, Pyankov SA, Sergeev AA. Enhancing the Protective Immune Response to Administration of a LIVP-GFP Live Attenuated Vaccinia Virus to Mice. Pathogens. 2021; 10(3):377. https://doi.org/10.3390/pathogens10030377
Chicago/Turabian StyleShchelkunov, Sergei N., Stanislav N. Yakubitskiy, Kseniya A. Titova, Stepan A. Pyankov, and Alexander A. Sergeev. 2021. "Enhancing the Protective Immune Response to Administration of a LIVP-GFP Live Attenuated Vaccinia Virus to Mice" Pathogens 10, no. 3: 377. https://doi.org/10.3390/pathogens10030377