
Exploring Prokaryotic and Eukaryotic Microbiomes Helps in Detecting Tick-Borne Infectious Agents in the Blood of Camels

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
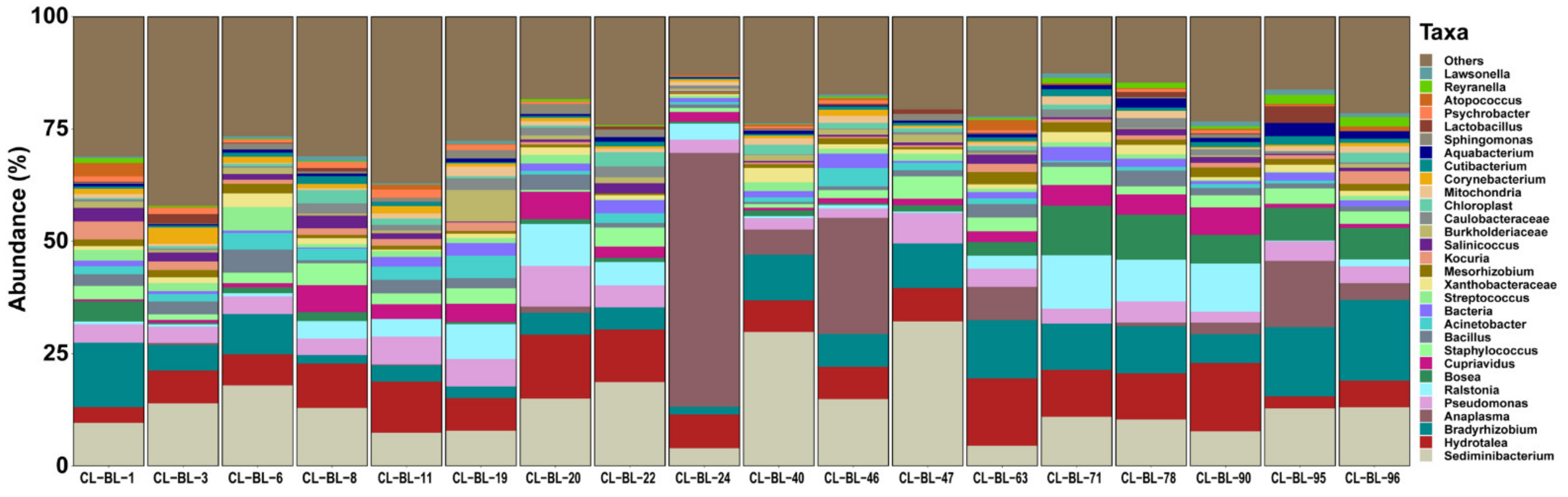
2.1. Bacterial Profile
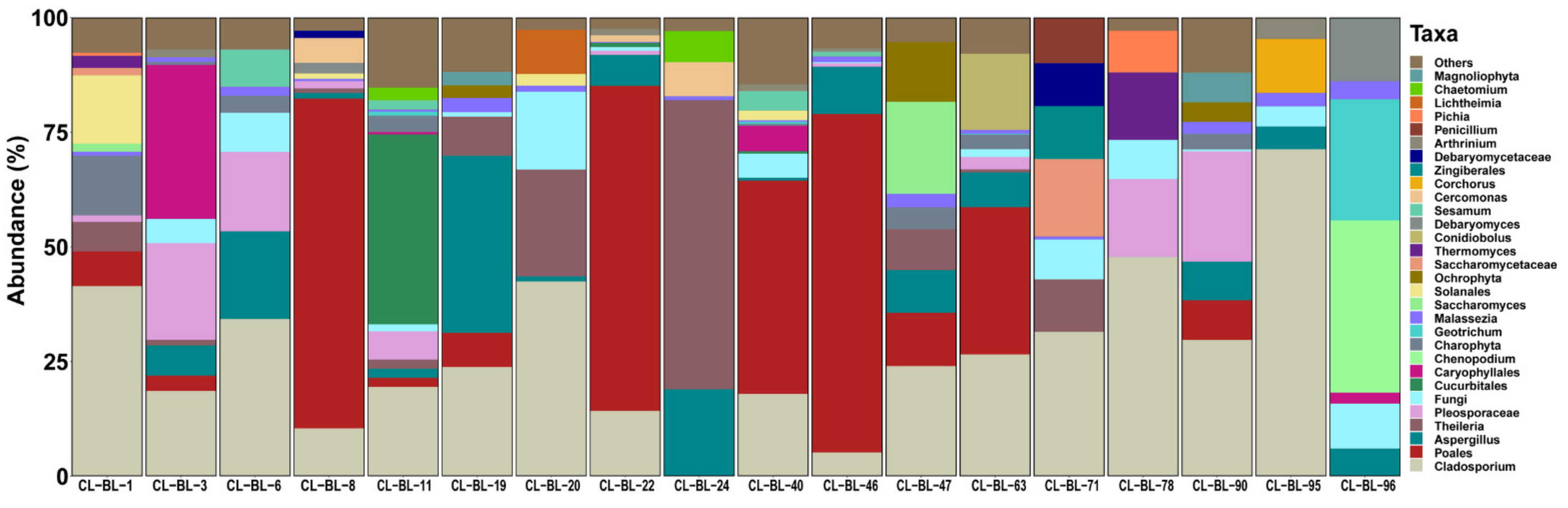
2.2. Eukaryotic Profile
2.3. Conventional Polymerase Chain Reaction (PCR) and Phylogenetic Relationships
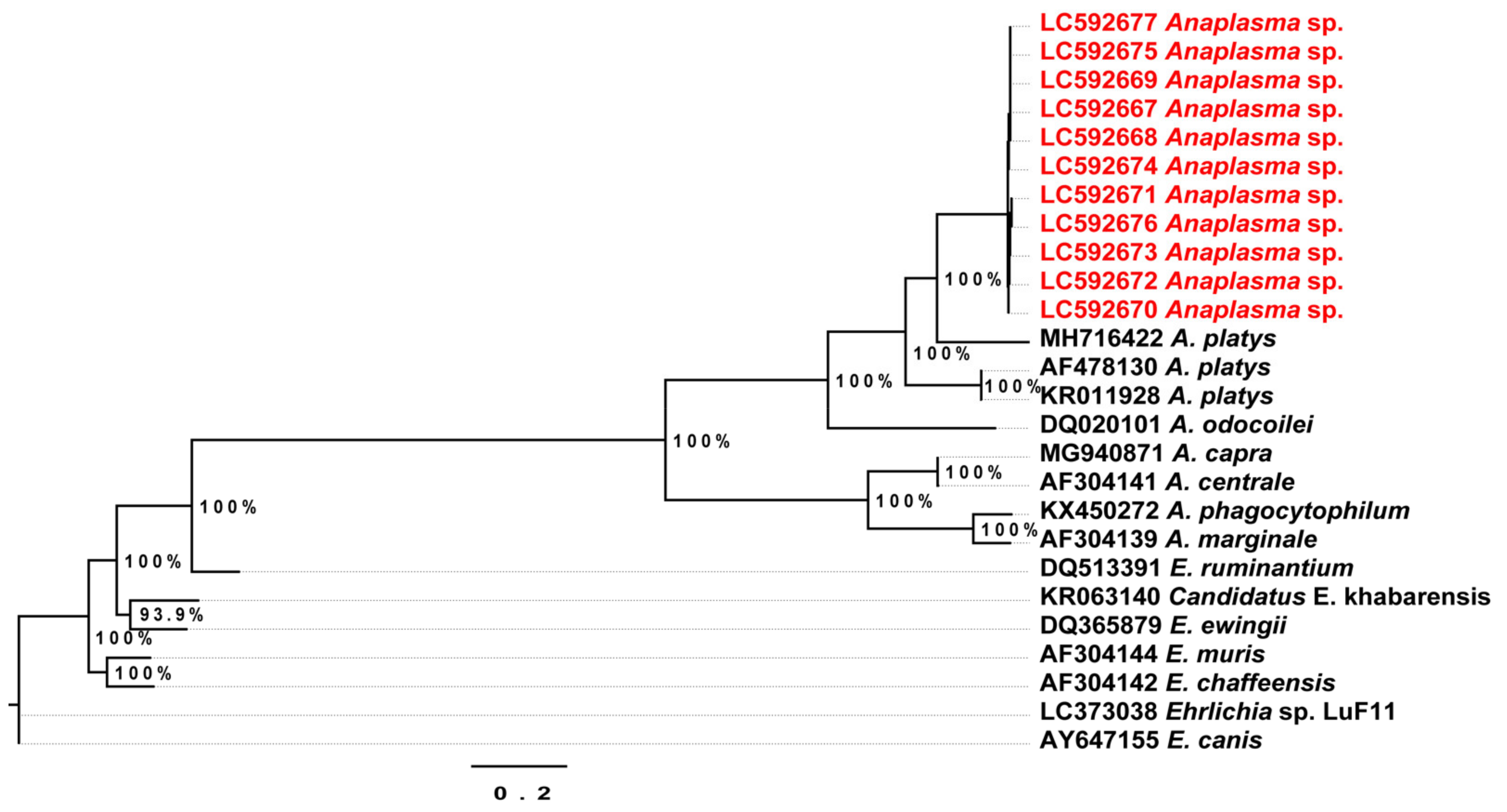
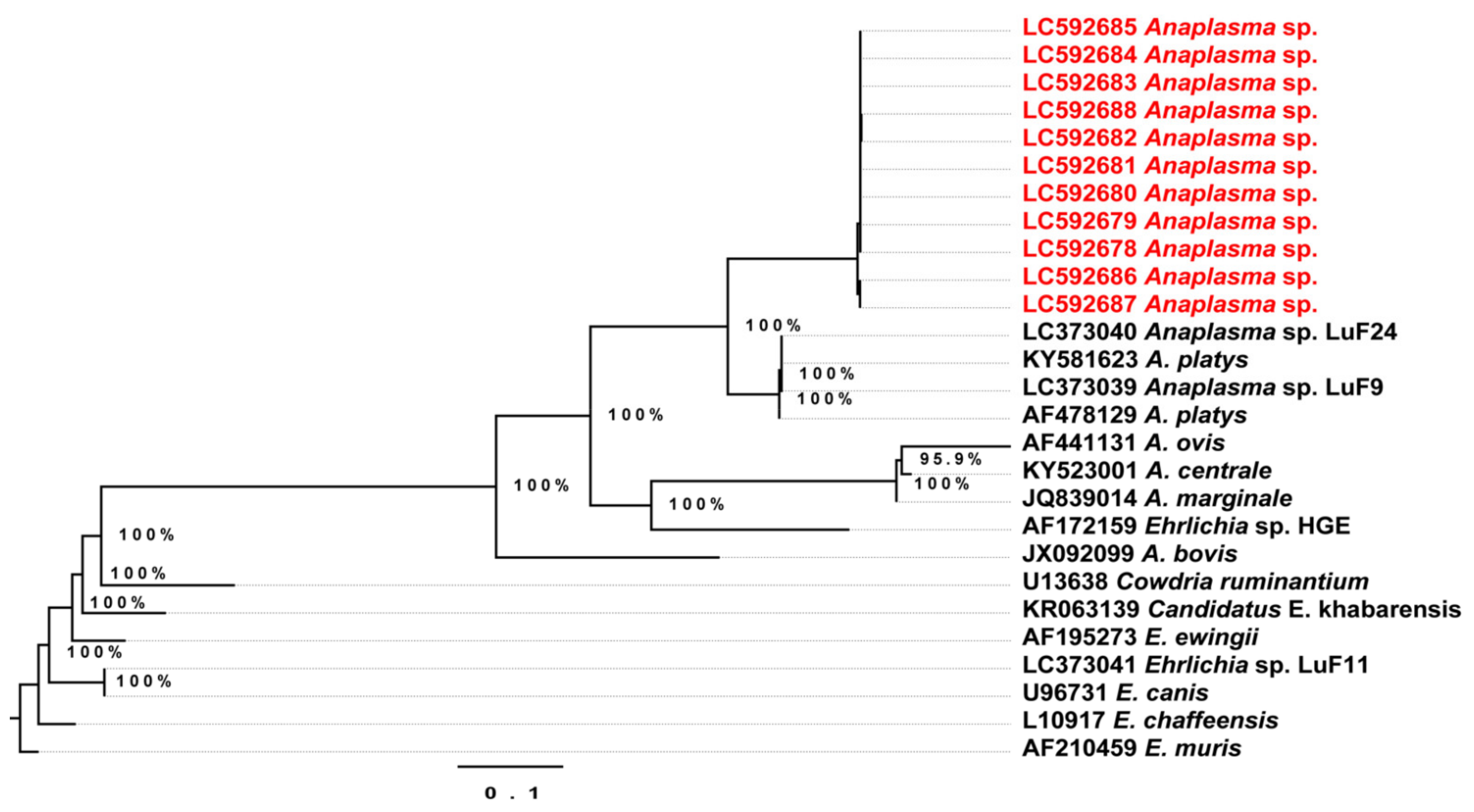
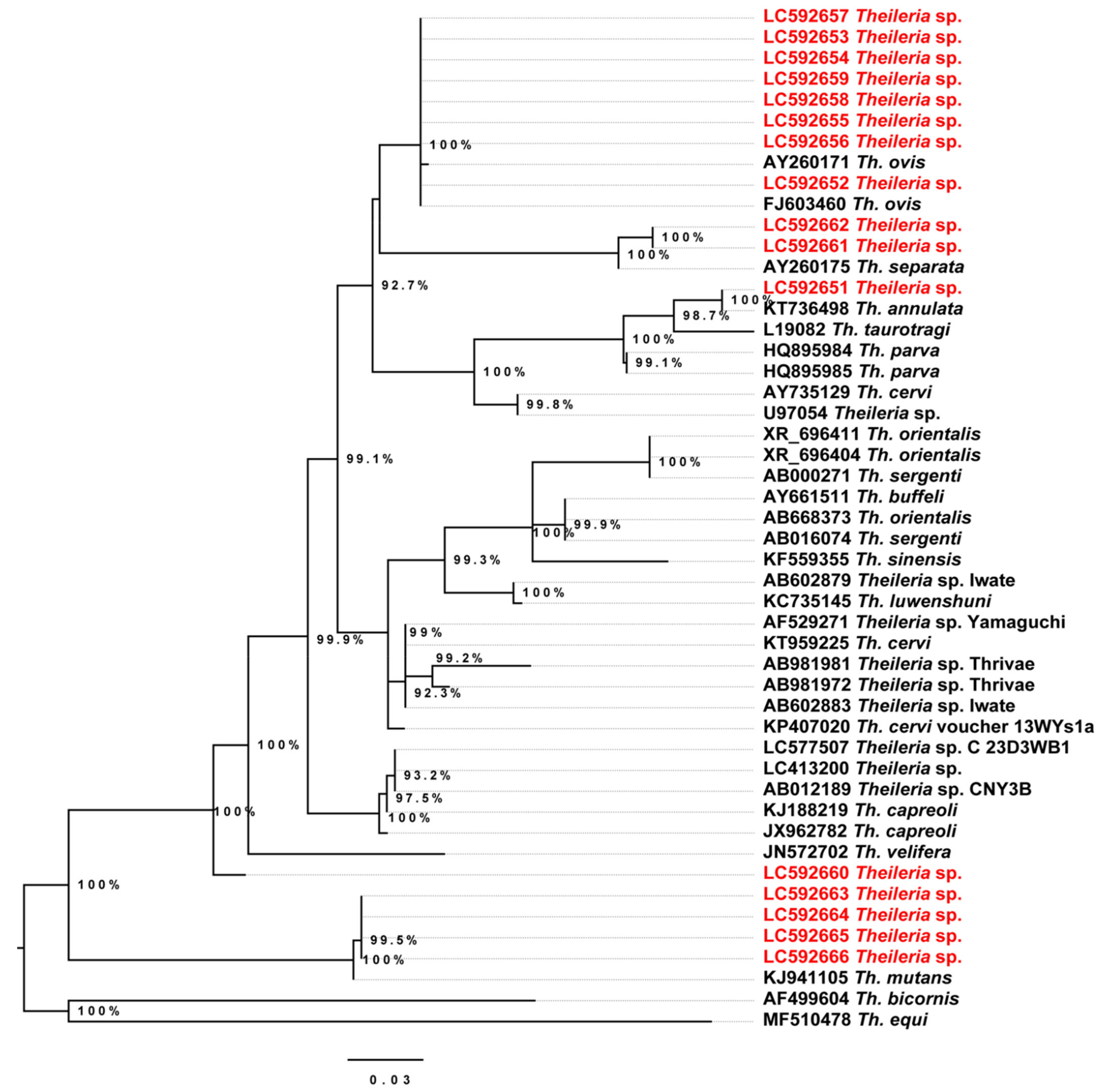
2.4. Phylogenetic Analysis of the Detected Theileria Species
2.5. Comparison of Next-Generation Sequencing (NGS)-Based and Conventional PCR Results
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Camel Blood Samples and DNA Extraction
5.2. 16S rDNA and 18S rDNA Amplification and Illumina MiSeq Sequencing
5.3. Illumina Data Processing
5.4. Conventional PCR and Sanger Sequencing
5.5. Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burger, P.A.; Ciani, E.; Faye, B. Old World camels in a modern world—A balancing act between conservation and genetic improvement. Anim. Genet. 2019, 50, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Chuluunbat, B.; Charruau, P.; Silbermayr, K.; Khorloojav, T.; Burger, P.A. Genetic diversity and population structure of Mongolian domestic Bactrian camels (Camelus bactrianus). Anim. Genet. 2014, 45, 550–558. [Google Scholar] [CrossRef] [Green Version]
- Alanazi, A.D.; Nguyen, V.L.; Alyousif, M.S.; Manoj, R.R.S.; Alouffi, A.S.; Donato, R.; Sazmand, A.; Mendoza-Roldan, J.A.; Dantas-Torres, F.; Otranto, D. Ticks and associated pathogens in camels (Camelus dromedarius) from Riyadh Province, Saudi Arabia. Parasites Vectors 2020, 13, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharbi, M.; Moussi, N.; Jedidi, M.; Mhadhbi, M.; Sassi, L.; Darghouth, M.A. Population dynamics of ticks infesting the one-humped camel (Camelus dromedarius) in central Tunisia. Ticks Tick-Borne Dis. 2013, 4, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Azmat, M.; Ijaz, M.; Farooqi, S.H.; Ghaffar, A.; Ali, A.; Masud, A.; Saleem, S.; Rehman, A.; Ali, M.M.; Mehmood, K.; et al. Molecular epidemiology, associated risk factors, and phylogenetic analysis of anaplasmosis in camel. Microb. Pathog. 2018, 123, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Bellabidi, M.; Benaissa, M.H.; Bissati-Bouafia, S.; Harrat, Z.; Brahmi, K.; Kernif, T. Coxiella burnetii in camels (Camelus dromedarius) from Algeria: Seroprevalence, molecular characterization, and ticks (Acari: Ixodidae) vectors. Acta Trop. 2020, 206, 105443. [Google Scholar] [CrossRef]
- Onyiche, T.E.; Raileanu, C.; Tauchmann, O.; Fischer, S.; Vasic, A.; Schafer, M.; Biu, A.A.; Ogo, N.I.; Thekisoe, O.; Silaghi, C. Prevalence and molecular characterization of ticks and tick-borne pathogens of one-humped camels (Camelus dromedarius) in Nigeria. Parasites Vectors 2020, 13, 428. [Google Scholar] [CrossRef]
- Selmi, R.; Ben Said, M.; Ben Yahia, H.; Abdelaali, H.; Messadi, L. Molecular epidemiology and phylogeny of spotted fever group Rickettsia in camels (Camelus dromedarius) and their infesting ticks from Tunisia. Transbound. Emerg. Dis. 2020, 67, 733–744. [Google Scholar] [CrossRef]
- Sazmand, A.; Eigner, B.; Mirzaei, M.; Hekmatimoghaddam, S.H.; Harl, J.; Duscher, G.G.; Fuehrer, H.; Joachim, A. Molecular identification of hemoprotozoan parasites in camels (Camelus dromedarius) of Iran. Iran. J. Parasitol. 2016, 11, 568–573. [Google Scholar] [PubMed]
- Youssef, S.Y.; Yasien, S.; Mousa, W.M.; Nasr, S.M.; El-Kelesh, E.A.; Mahran, K.M.; Abd-El-Rahman, A.H. Vector identification and clinical, hematological, biochemical, and parasitological characteristics of camel (Camelus dromedarius) theileriosis in Egypt. Trop. Anim. Health Prod. 2015, 47, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.; Pons, M.J.; Herranz, M.; Wakeman, K.C.; Del Valle, J.; Vermeij, M.J.A.; Leander, B.S.; Keeling, P.J. Validation of a universal set of primers to study animal-associated microeukaryotic communities. Environ. Microbiol. 2019, 21, 3855–3861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstock, G.M.; Goldberg, B.; Ledeboer, N.; Rubin, E.; Sichtig, H.; Geyer, C. Applications of Clinical Microbial Next-Generation Sequencing. Am. Soc. Microbiol. 2016. [Google Scholar] [CrossRef]
- Wilcox, J.J.S.; Hollocher, H. Unprecedented symbiont eukaryote diversity is governed by internal trophic webs in a wild non-human primate. Protist 2018, 169, 307–320. [Google Scholar] [CrossRef]
- Carnegie, R.B.; Meyer, G.R.; Blackbourn, J.; Cochennec-Laureau, N.; Berthe, F.C.; Bower, S.M. Molecular detection of the oyster parasite Mikrocytos mackini, and a preliminary phylogenetic analysis. Dis. Aquat. Org. 2003, 54, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Ondrejicka, D.A.; Locke, S.A.; Morey, K.; Borisenko, A.V.; Hanner, R.H. Status and prospects of DNA barcoding in medically important parasites and vectors. Trends Parasitol. 2014, 30, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Huggins, L.G.; Koehler, A.V.; Ng-Nguyen, D.; Wilcox, S.; Schunack, B.; Inpankaew, T.; Traub, R.J. A novel metabarcoding diagnostic tool to explore protozoan haemoparasite diversity in mammals: A proof-of-concept study using canines from the tropics. Sci. Rep. 2019, 9, 12644. [Google Scholar] [CrossRef] [Green Version]
- Ellis, J.E.; Missan, D.S.; Shabilla, M.; Moschonas, C.; Saperstein, D.; Martinez, D.; Becker, C.V.; Fry, S.E. Comparison of the prokaryotic and eukaryotic microbial communities in peripheral blood from amyotrophic lateral sclerosis, multiple sclerosis, and control populations. Hum. Microbiome J. 2019, 13, 100060. [Google Scholar] [CrossRef]
- Castillo, D.J.; Rifkin, R.F.; Cowan, D.A.; Potgieter, M. The healthy human blood microbiome: Fact or fiction? Front. Cell. Infect. Microbiol. 2019, 9, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sze, M.A.; Tsuruta, M.; Yang, S.W.; Oh, Y.; Man, S.F.; Hogg, J.C.; Sin, D.D. Changes in the bacterial microbiota in gut, blood, and lungs following acute LPS instillation into mice lungs. PLoS ONE 2014, 9, e111228. [Google Scholar] [CrossRef] [Green Version]
- Vientos-Plotts, A.I.; Ericsson, A.C.; Rindt, H.; Grobman, M.E.; Graham, A.; Bishop, K.; Cohn, L.A.; Reinero, C.R. Dynamic changes of the respiratory microbiota and its relationship to fecal and blood microbiota in healthy young cats. PLoS ONE 2017, 12, e0173818. [Google Scholar] [CrossRef] [PubMed]
- Paisse, S.; Valle, C.; Servant, F.; Courtney, M.; Burcelin, R.; Amar, J.; Lelouvier, B. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 2016, 56, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Napp, S.; Chevalier, V.; Busquets, N.; Calistri, P.; Casal, J.; Attia, M.; Elbassal, R.; Hosni, H.; Farrag, H.; Hassan, N.; et al. Understanding the legal trade of cattle and camels and the derived risk of Rift Valley Fever introduction into and transmission within Egypt. PLoS Negl. Trop. Dis. 2018, 12, e0006143. [Google Scholar] [CrossRef] [Green Version]
- Ahmed Kamal, S. Observations on rift valley fever virus and vaccines in Egypt. Virol. J. 2011, 8, 532. [Google Scholar] [CrossRef] [Green Version]
- Chu, D.K.; Poon, L.L.; Gomaa, M.M.; Shehata, M.M.; Perera, R.A.; Abu Zeid, D.; El Rifay, A.S.; Siu, L.Y.; Guan, Y.; Webby, R.J.; et al. MERS coronaviruses in dromedary camels, Egypt. Emerg. Infect. Dis. 2014, 20, 1049–1053. [Google Scholar] [CrossRef] [Green Version]
- Song, S.J.; Sanders, J.G.; Delsuc, F.; Metcalf, J.; Amato, K.; Taylor, M.W.; Mazel, F.; Lutz, H.L.; Winker, K.; Graves, G.R.; et al. Comparative analyses of vertebrate gut microbiomes reveal convergence between birds and bats. MBio 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penders, J.; Thijs, C.; Vink, C.; Stelma, F.F.; Snijders, B.; Kummeling, I.; Van den Brandt, P.A.; Stobberingh, E.E. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 2006, 118, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Whittle, E.; Leonard, M.O.; Harrison, R.; Gant, T.W.; Tonge, D.P. Multi-method characterization of the human circulating microbiome. Front. Microbiol. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narasimhan, S.; Rajeevan, N.; Liu, L.; Zhao, Y.O.; Heisig, J.; Pan, J.; Eppler-Epstein, R.; Deponte, K.; Fish, D.; Fikrig, E. Gut microbiota of the tick vector Ixodes scapularis modulate colonization of the Lyme disease spirochete. Cell Host Microbe 2014, 15, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Laroche, M.; Raoult, D.; Parola, P. Insects and the transmission of bacterial agents. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef]
- El Imam, A.H.; Hassan, S.M.; Gameel, A.A.; El Hussein, A.M.; Taha, K.M.; Oosthuizen, M.C. Molecular identification of different Theileria and Babesia species infecting sheep in Sudan. Ann. Parasitol. 2016, 62, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Magzoub, A.; El Ghali, A.; Hussien, M.O.; Juma, Y.; Mohammed, S.B. Prevalence of ticks (Acari: Ixodidae) and Theileria lestoquardi in sheep at El Huda and El Nuhud animals production research stations, Sudan. J. Parasit. Dis. 2020. [Google Scholar] [CrossRef]
- Springer, A.; Shuaib, Y.A.; Isaa, M.H.; Ezz-Eldin, M.I.; Osman, A.Y.; Yagoub, I.A.; Abdalla, M.A.; Bakiet, A.O.; Mohmed-Noor, S.E.; Schaper, S.; et al. Tick fauna and associated Rickettsia, Theileria, and Babesia spp. in domestic animals in Sudan (north Kordofan and Kassala states). Microorganisms 2020, 8, 1969. [Google Scholar] [CrossRef] [PubMed]
- Shemshad, M.; Shemshad, K.; Sedaghat, M.M.; Shokri, M.; Barmaki, A.; Baniardalani, M.; Rafinejad, J. First survey of hard ticks (Acari: Ixodidae) on cattle, sheep and goats in Boeen Zahra and Takistan counties, Iran. Asian Pac. J. Trop. Biomed. 2012, 2, 489–492. [Google Scholar] [CrossRef] [Green Version]
- Bouattour, A.; Darghouth, M.A.; Daoud, A. Distribution and ecology of ticks (Acari: Ixodidae) infesting livestock in Tunisia: An overview of eighth years field collections. Parassitologia 1999, 41 (Suppl. 1), 5–10. [Google Scholar]
- Elsify, A.; Sivakumar, T.; Nayel, M.; Salama, A.; Elkhtam, A.; Rizk, M.; Mosaab, O.; Sultan, K.; Elsayed, S.; Igarashi, I.; et al. An epidemiological survey of bovine Babesia and Theileria parasites in cattle, buffaloes, and sheep in Egypt. Parasitol. Int. 2015, 64, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Tomassone, L.; Grego, E.; Callà, G.; Rodighiero, P.; Pressi, G.; Gebre, S.; Zeleke, B.; De Meneghi, D. Ticks and tick-borne pathogens in livestock from nomadic herds in the Somali Region, Ethiopia. Exp. Appl. Acarol. 2012, 56, 391–401. [Google Scholar] [CrossRef]
- Qablan, M.A.; Sloboda, M.; Jirků, M.; Oborník, M.; Dwairi, S.; Amr, Z.S.; Hořín, P.; Lukeš, J.; Modrý, D. Quest for the piroplasms in camels: Identification of Theileria equi and Babesia caballi in Jordanian dromedaries by PCR. Vet. Parasitol. 2012, 186, 456–460. [Google Scholar] [CrossRef]
- Lorusso, V.; Wijnveld, M.; Latrofa, M.S.; Fajinmi, A.; Majekodunmi, A.O.; Dogo, A.G.; Igweh, A.C.; Otranto, D.; Jongejan, F.; Welburn, S.C.; et al. Canine and ovine tick-borne pathogens in camels, Nigeria. Vet. Parasitol. 2016, 228, 90–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glidden, C.K.; Koehler, A.V.; Hall, R.S.; Saeed, M.A.; Coppo, M.; Beechler, B.R.; Charleston, B.; Gasser, R.B.; Jolles, A.E.; Jabbar, A. Elucidating cryptic dynamics of Theileria communities in African buffalo using a high-throughput sequencing informatics approach. Ecol. Evol. 2020, 10, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squarre, D.; Nakamura, Y.; Hayashida, K.; Kawai, N.; Chambaro, H.; Namangala, B.; Sugimoto, C.; Yamagishi, J. Investigation of the piroplasm diversity circulating in wildlife and cattle of the greater Kafue ecosystem, Zambia. Parasites Vectors 2020, 13, 599. [Google Scholar] [CrossRef]
- Amer, S.; Ryu, O.; Tada, C.; Fukuda, Y.; Inoue, N.; Nakai, Y. Molecular identification and phylogenetic analysis of Trypanosoma evansi from dromedary camels (Camelus dromedarius) in Egypt, a pilot study. Acta Trop. 2011, 117, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Elhaig, M.M.; Sallam, N.H. Molecular survey and characterization of Trypanosoma evansi in naturally infected camels with suspicion of a Trypanozoon infection in horses by molecular detection in Egypt. Microb. Pathog. 2018, 123, 201–205. [Google Scholar] [CrossRef]
- Bastos, A.D.; Mohammed, O.B.; Bennett, N.C.; Petevinos, C.; Alagaili, A.N. Molecular detection of novel Anaplasmataceae closely related to Anaplasma platys and Ehrlichia canis in the dromedary camel (Camelus dromedarius). Vet. Microbiol. 2015, 179, 310–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidambasi, K.O.; Masiga, D.K.; Villinger, J.; Carrington, M.; Bargul, J.L. Detection of blood pathogens in camels and their associated ectoparasitic camel biting keds, Hippobosca camelina: The potential application of keds in xenodiagnosis of camel haemopathogens. AAS Open Res. 2019, 2, 164. [Google Scholar] [CrossRef]
- Ait Lbacha, H.; Zouagui, Z.; Alali, S.; Rhalem, A.; Petit, E.; Ducrotoy, M.J.; Boulouis, H.J.; Maillard, R. “Candidatus anaplasma camelii” in one-humped camels (Camelus dromedarius) in Morocco: A novel and emerging anaplasma species? Infect. Dis. Poverty 2017, 6, 1. [Google Scholar] [CrossRef]
- Sharifiyazdi, H.; Jafari, S.; Ghane, M.; Nazifi, S.; Sanati, A. Molecular investigation of Anaplasma and Ehrlichia natural infections in the dromedary camel (Camelus dromedarius) in Iran. Comp. Clin. Pathol. 2017, 26, 99–103. [Google Scholar] [CrossRef]
- Koh, F.X.; Panchadcharam, C.; Sitam, F.T.; Tay, S.T. Molecular investigation of Anaplasma spp. in domestic and wildlife animals in Peninsular Malaysia. Vet. Parasitol. Reg. Stud. Rep. 2018, 13, 141–147. [Google Scholar] [CrossRef]
- Parvizi, O.; El-Adawy, H.; Roesler, U.; Neubauer, H.; Mertens-Scholz, K. Performance analysis of Anaplasma antibody competitive ELISA using the ROC curve for screening of anaplasmosis in camel populations in Egypt. Pathogens 2020, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Qi, R.J.; Jiang, J.Z.; Zhang, M.Q.; Wang, J.Y. Development of a blocking primer to inhibit the PCR amplification of the 18S rDNA Sequences of Litopenaeus vannamei and its efficacy in Crassostrea hongkongensis. Front. Microbiol. 2019, 10, 830. [Google Scholar] [CrossRef] [PubMed]
- Belda, E.; Coulibaly, B.; Fofana, A.; Beavogui, A.H.; Traore, S.F.; Gohl, D.M.; Vernick, K.D.; Riehle, M.M. Preferential suppression of Anopheles gambiae host sequences allows detection of the mosquito eukaryotic microbiome. Sci. Rep. 2017, 7, 3241. [Google Scholar] [CrossRef] [PubMed]
- Reigel, A.M.; Owens, S.M.; Hellberg, M.E. Reducing host DNA contamination in 16S rRNA gene surveys of anthozoan microbiomes using PNA clamps. Coral Reefs 2020, 39, 1817–1827. [Google Scholar] [CrossRef]
- Jian, C.; Luukkonen, P.; Yki-Järvinen, H.; Salonen, A.; Korpela, K. Quantitative PCR provides a simple and accessible method for quantitative microbiota profiling. PLoS ONE 2020, 15, e0227285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herlemann, D.P.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Matjila, P.T.; Penzhorn, B.L.; Bekker, C.P.; Nijhof, A.M.; Jongejan, F. Confirmation of occurrence of Babesia canis vogeli in domestic dogs in South Africa. Vet. Parasitol. 2004, 122, 119–125. [Google Scholar] [CrossRef]
- Njiru, Z.K.; Constantine, C.C.; Guya, S.; Crowther, J.; Kiragu, J.M.; Thompson, R.C.; Davila, A.M. The use of ITS1 rDNA PCR in detecting pathogenic African trypanosomes. Parasitol. Res. 2005, 95, 186–192. [Google Scholar] [CrossRef]
- Parola, P.; Roux, V.; Camicas, J.L.; Baradji, I.; Brouqui, P.; Raoult, D. Detection of ehrlichiae in African ticks by polymerase chain reaction. Trans. R. Soc. Trop. Med. Hyg. 2000, 94, 707–708. [Google Scholar] [CrossRef]
- Inokuma, H.; Brouqui, P.; Drancourt, M.; Raoult, D. Citrate synthase gene sequence: A new tool for phylogenetic analysis and identification of Ehrlichia. J. Clin. Microbiol. 2001, 39, 3031–3039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumner, J.W.; Nicholson, W.L.; Massung, R.F. PCR amplification and comparison of nucleotide sequences from the groESL heat shock operon of Ehrlichia species. J. Clin. Microbiol. 1997, 35, 2087–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liz, J.S.; Anderes, L.; Sumner, J.W.; Massung, R.F.; Gern, L.; Rutti, B.; Brossard, M. PCR detection of granulocytic ehrlichiae in Ixodes ricinus ticks and wild small mammals in western Switzerland. J. Clin. Microbiol. 2000, 38, 1002–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gofton, A.W.; Doggett, S.; Ratchford, A.; Ryan, U.; Irwin, P. Phylogenetic characterisation of two novel Anaplasmataceae from Australian Ixodes holocyclus ticks: ‘Candidatus Neoehrlichia australis’ and ‘Candidatus Neoehrlichia arcana’. Int. J. Syst. Evol. Microbiol. 2016, 66, 4256–4261. [Google Scholar] [CrossRef]
- Urakawa, T.; Verloo, D.; Moens, L.; Buscher, P.; Majiwa, P.A. Trypanosoma evansi: Cloning and expression in Spodoptera frugiperda [correction of fugiperda] insect cells of the diagnostic antigen RoTat1.2. Exp. Parasitol. 2001, 99, 181–189. [Google Scholar] [CrossRef]
- Njiru, Z.K.; Constantine, C.C.; Masiga, D.K.; Reid, S.A.; Thompson, R.C.; Gibson, W.C. Characterization of Trypanosoma evansi type B. Infect. Genet. Evol. 2006, 6, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Bower, S.M.; Carnegie, R.B.; Goh, B.; Jones, S.R.; Lowe, G.J.; Mak, M.W. Preferential PCR amplification of parasitic protistan small subunit rDNA from metazoan tissues. J. Eukaryot. Microbiol. 2004, 51, 325–332. [Google Scholar] [CrossRef]
- Comeau, A.M.; Li, W.K.; Tremblay, J.E.; Carmack, E.C.; Lovejoy, C. Arctic Ocean microbial community structure before and after the 2007 record sea ice minimum. PLoS ONE 2011, 6, e27492. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Kaneko, C.; Kajihara, M.; Ngonda, S.; Simulundu, E.; Muleya, W.; Thu, M.J.; Hang’ombe, M.B.; Katakura, K.; Takada, A.; et al. Tick-borne haemoparasites and Anaplasmataceae in domestic dogs in Zambia. Ticks Tick-Borne Dis. 2018, 9, 988–995. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Candidatus Anaplasma Camelii | Theileria annulata | Theileria ovis | Theileria separata | Theileria mutans-Like * | Theileria sp. |
|---|---|---|---|---|---|---|
| CL-BL-1 | 28 | 0 | 488 (LC592658) ** | 0 | 241 (LC592666) | 65 (LC592660) |
| CL-BL-3 | 0 | 0 | 171 (LC592657) | 0 | 0 | 0 |
| CL-BL-6 | 0 | 0 | 0 | 0 | 0 | 0 |
| CL-BL-8 | 324 | 0 | 175 (LC592656) | 0 | 0 | 0 |
| CL-BL-11 | 0 | 0 | 120 (LC592659) | 0 | 0 | 0 |
| CL-BL-19 | 13,101 | 314 (LC592651) | 434 (LC592655) | 0 | 0 | 0 |
| CL-BL-20 | 128 | 0 | 1252 (LC592654) | 0 | 572 (LC592665) | 0 |
| CL-BL-22 | 931 | 0 | 0 | 0 | 0 | 0 |
| CL-BL-24 | 1914 | 0 | 2945 (LC592653) | 626 (LC592662) | 1360 (LC592664) | 0 |
| CL-BL-40 | 0 | 0 | 0 | 0 | 0 | 0 |
| CL-BL-46 | 909 | 0 | 0 | 0 | 0 | 0 |
| CL-BL-47 | 0 | 0 | 0 | 558 (LC592661) | 0 | 0 |
| CL-BL-63 | 0 | 0 | 0 | 0 | 77 (LC592663) | 0 |
| CL-BL-71 | 88 | 0 | 513 (LC592652) | 0 | 0 | 0 |
| CL-BL-78 | 0 | 0 | 0 | 0 | 0 | 0 |
| CL-BL-90 | 461 | 0 | 0 | 0 | 0 | 0 |
| CL-BL-95 | 2738 | 0 | 0 | 0 | 0 | 0 |
| CL-BL-96 | 589 | 0 | 0 | 0 | 0 | 0 |
| Sample ID | Anaplasma spp. | Theileria spp. | Trypanosoma evansi | |||||
|---|---|---|---|---|---|---|---|---|
| NGS (V3-V4-PCR) | EHR-PCR | gltA-PCR | groEL-PCR | NGS (UNonMet-PCR) | RLB-PCR | NGS (UNonMet-PCR) | ILO-PCR | |
| CL-BL-1 | N | N | N | N | P | N | N | N |
| CL-BL-3 | P | N | N | N | P | N | N | N |
| CL-BL-6 | N | N | N | N | N | N | N | N |
| CL-BL-8 | N | N | N | N | P | N | N | P |
| CL-BL-11 | P | N | N | N | P | N | N | N |
| CL-BL-19 | N | N | N | N | P | N | N | P |
| CL-BL-20 | P | N | N | N | P | N | N | N |
| CL-BL-22 | N | N | N | N | N | N | N | N |
| CL-BL-24 | P | N | N | N | P | N | N | N |
| CL-BL-40 | P | P | P | P | N | N | N | P |
| CL-BL-46 | P | P | P | P | N | N | N | P |
| CL-BL-47 | N | N | N | N | P | N | N | P |
| CL-BL-63 | P | N | N | N | P | N | N | N |
| CL-BL-71 | N | N | N | N | P | N | N | N |
| CL-BL-78 | P | N | N | N | N | N | N | N |
| CL-BL-90 | P | P | P | P | N | N | N | P |
| CL-BL-95 | P | P | P | P | N | N | N | N |
| CL-BL-96 | P | P | P | P | N | N | N | N |
| PCR Name | Primer Name | Primer Sequence (5′–3′) | Annealing Temp/Extension Time | Reference |
|---|---|---|---|---|
| V3-V4-PCR | Illumina_16S_341F | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG | 55 °C/30 s | [54] |
| Illumina_16S_805R | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC | |||
| UNonMet-PCR | 18S-EUK581-F | GTGCCAGCAGCCGCG | 62 °C/30 s | [22] |
| 18S-EUK1134-R | TTTAAGTTTCAGCCTTGCG | |||
| V4-PCR | Illumina_E572F | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCYGCGGTAATTCCAGCTC | 55 °C/30 s | [23] |
| Illumina_E1009R | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGAYGGTATCTRATCRTCTTYG | |||
| RLB-PCR | RLB-F2 | GACACAGGGAGGTAGTGACAAG | 54 °C/15 s | [55] |
| RLB-R2 | CTAAGAATTTCACCTCTGACAGT | |||
| ITS1-PCR | ITS1-CF | CCGGAAGTTCACCGATATTG | 52 °C/15 s | [56] |
| ITS1-BR | TTGCTGCGTTCTTCAACGAA | |||
| EHR-PCR | EHR16SD | GGTACCYACAGAAGAAGTCC | 61 °C/15 s | [57] |
| EHR16SR | TAGCACTCATCGTTTACAGC | |||
| gltA-PCR (1st) | F4b | CCGGGTTTTATGTCTACTGC | 55 °C/15 s | [58] |
| R1b | CGATGACCAAAACCCAT | |||
| gltA-PCR (2nd) | EHR-CS136F | TTYATGTCYACTGCTGCKTG | 50 °C/15 s | [58] |
| EHR-CS778R | GCNCCMCCATGMGCTGG | |||
| groEL-PCR (1st) | HS1-F | CGYCAGTGGGCTGGTAATGAA | 54 °C/15 s | [59,60] |
| HS6-R | CCWCCWGGTACWACACCTTC | |||
| groEL-PCR (2nd) | HS3-F | ATAGTYATGAAGGAGAGTGAT | 50 °C/15 s | [60,61] |
| HSV-R | TCAACAGCAGCTCTAGTWG | |||
| ILO-PCR | ILO7957 | GCCACCACGGCGAAAGAC | 52 °C/15 s | [62] |
| ILO8091 | TAATCAGTGTGGTGTGC | |||
| EVAB-PCR | EVAB1 | CACAGTCCGAGAGATAGAG | 60 °C/15 s | [63] |
| EVAB2 | CTGTACTCTACATCTACCTC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, W.M.A.; Ali, A.O.; Mahmoud, H.Y.A.H.; Omar, M.A.; Chatanga, E.; Salim, B.; Naguib, D.; Anders, J.L.; Nonaka, N.; Moustafa, M.A.M.; et al. Exploring Prokaryotic and Eukaryotic Microbiomes Helps in Detecting Tick-Borne Infectious Agents in the Blood of Camels. Pathogens 2021, 10, 351. https://doi.org/10.3390/pathogens10030351
Mohamed WMA, Ali AO, Mahmoud HYAH, Omar MA, Chatanga E, Salim B, Naguib D, Anders JL, Nonaka N, Moustafa MAM, et al. Exploring Prokaryotic and Eukaryotic Microbiomes Helps in Detecting Tick-Borne Infectious Agents in the Blood of Camels. Pathogens. 2021; 10(3):351. https://doi.org/10.3390/pathogens10030351
Chicago/Turabian StyleMohamed, Wessam Mohamed Ahmed, Alsagher O. Ali, Hassan Y. A. H. Mahmoud, Mosaab A. Omar, Elisha Chatanga, Bashir Salim, Doaa Naguib, Jason L. Anders, Nariaki Nonaka, Mohamed Abdallah Mohamed Moustafa, and et al. 2021. "Exploring Prokaryotic and Eukaryotic Microbiomes Helps in Detecting Tick-Borne Infectious Agents in the Blood of Camels" Pathogens 10, no. 3: 351. https://doi.org/10.3390/pathogens10030351