
A Snapshot of the Genetic Diversity of Salmonella Enteritidis Population Involved in Human Infections in Romania Taken in the European Epidemiological Context

 ,
,
Abstract
:1. Introduction
2. Results
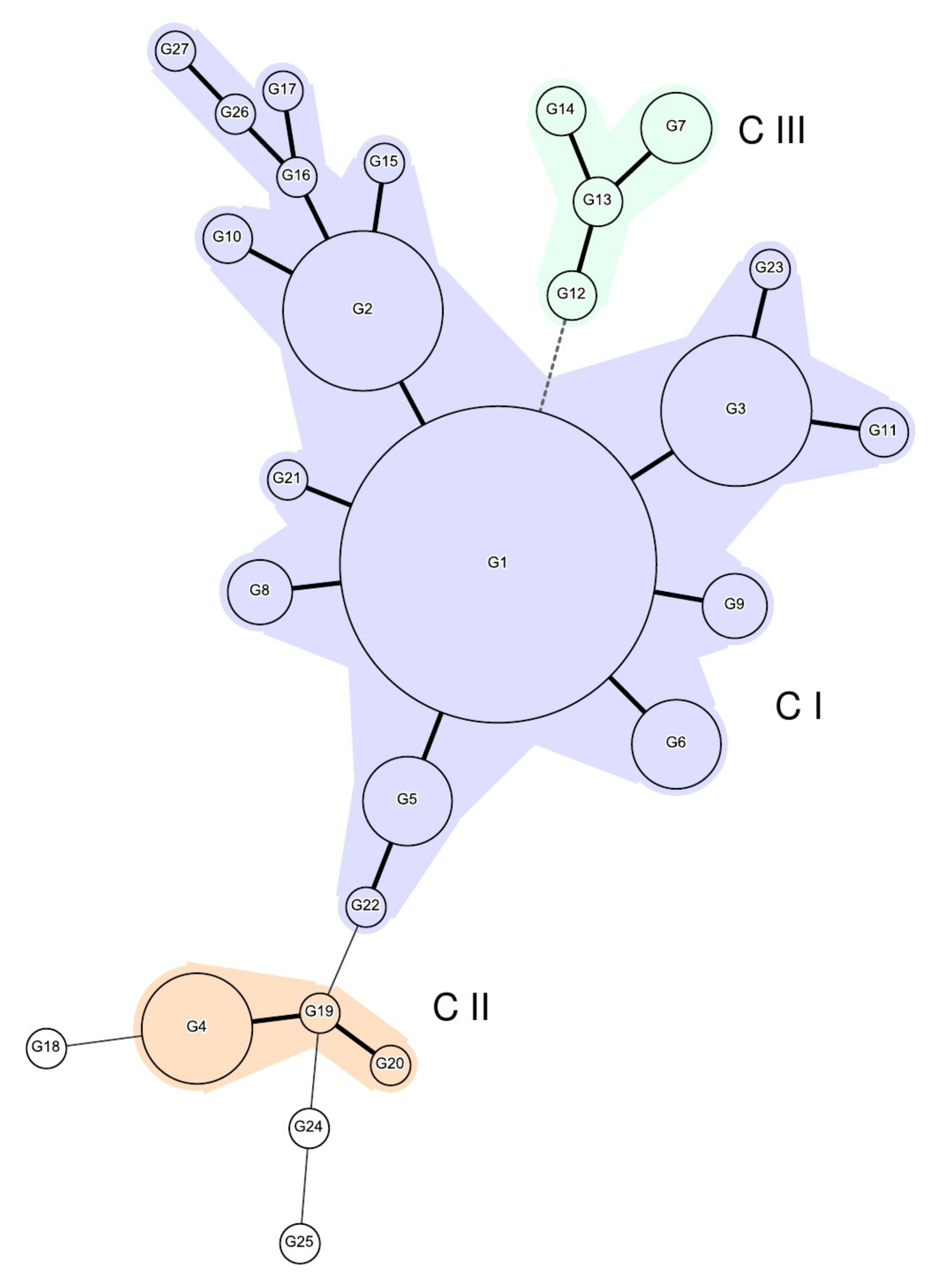
2.1. MLVA Typing
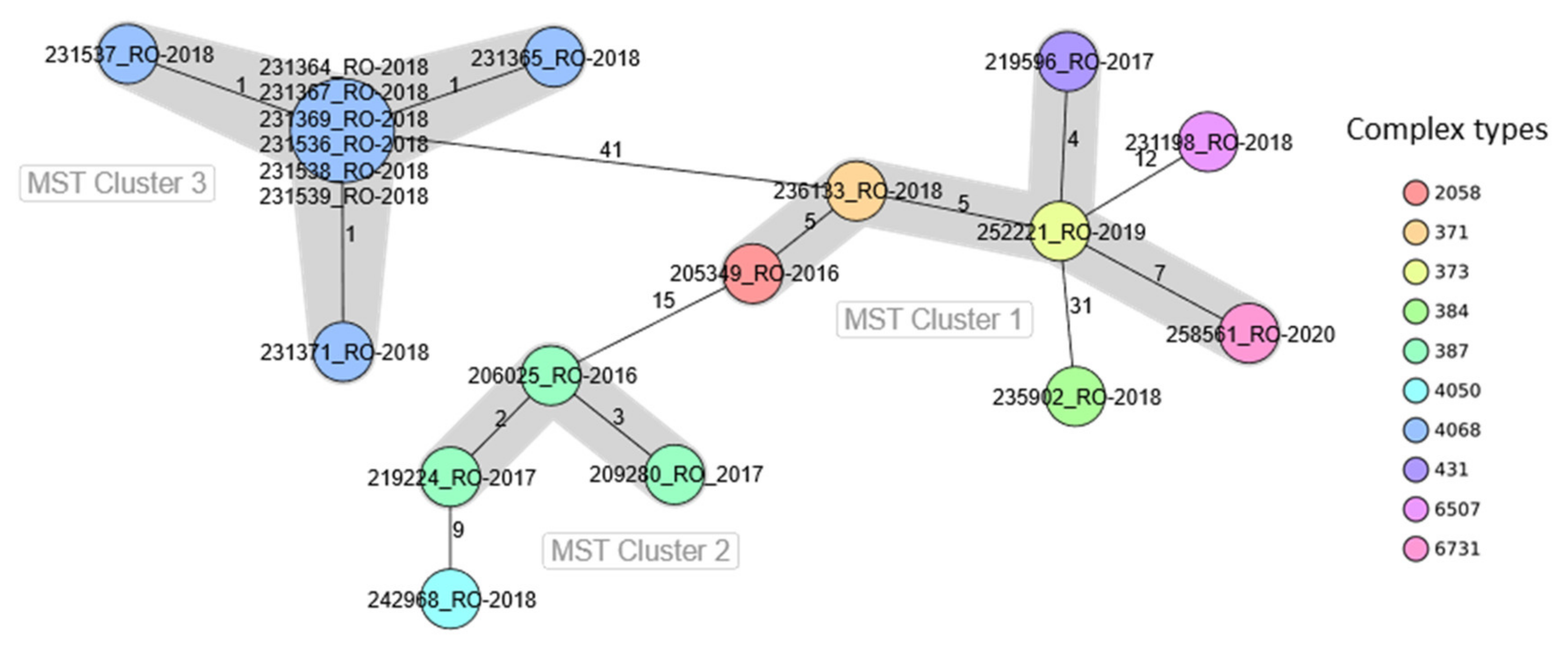
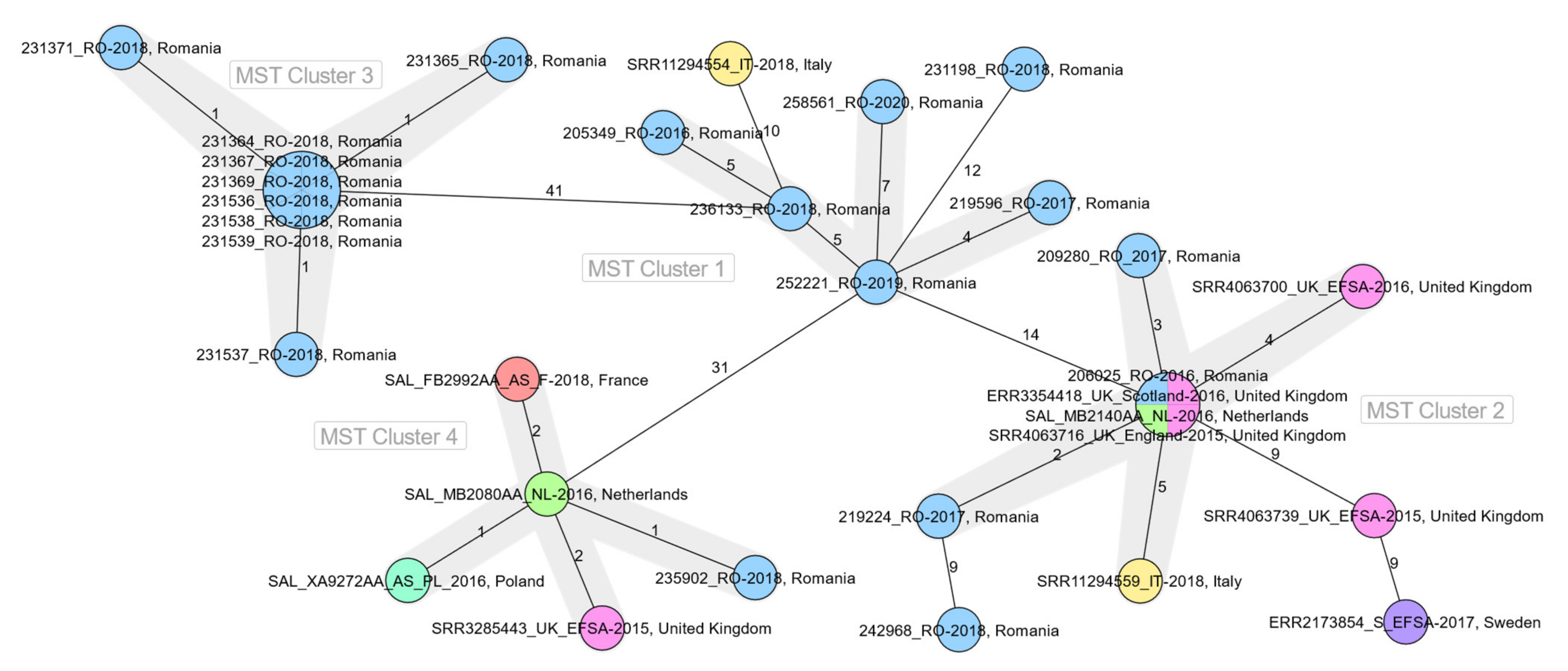
2.2. WGS-Based Typing
2.3. In Vitro and In Silico Antibiotic Resistance in Relationship with the MLVA Type
3. Discussion
4. Methods
4.1. Bacterial Strains
4.2. Multiple-Locus Variable-Number of Tandem Repeats Analysis (MLVA)
4.3. Whole Genome Sequencing
4.4. In Vitro Antimicrobial Susceptibility Testing
4.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AB | Alba |
| BN | Bistriţa-Năsăud |
| BT | Botoşani |
| BR | Brăila |
| BV | Braşov |
| B | Bucharest |
| BZ | Buzău |
| CL | Călăraşi |
| CT | Constanţa |
| CV | Covasna |
| DB | Dâmboviţa |
| HR | Harghita |
| IS | Iaşi |
| MM | Maramures |
| MH | Mehedinţi |
| OT | Olt |
| PH | Prahova |
| SM | Satu Mare |
| SV | Suceava |
| TM | Timiş |
| TL | Tulcea |
| VS | Vaslui |
| VN | Vrancea |
| AMP | Ampicillin |
| CIP | Ciprofloxacin |
| NA | Nalidixic acid |
| PEF | Pefloxacin |
| S | Streptomycin |
| S3 | Sulphonamides |
| R | Resistant |
References
- European Food Safety Authority; European Centre for Disease Prevention and Control. The European Union One Health 2019 Zoonoses Report. EFSA J. 2021, 19, e06406. [Google Scholar] [CrossRef]
- Kruy, S.L.; Cuyck, H.; Koeck, J.L. Multilocus variable number tandem repeat analysis for Salmonella enterica subspecies. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30, 465–473. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control. ECDC Strategic Framework for the Integration of Molecular and Genomic Typing into European Surveillance and Multi-Country Outbreak Investigations—2019–2021. 2019. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/framework-for-genomic-surveillance.pdf (accessed on 11 October 2021).
- European Centre for Disease Prevention and Control. EU Laboratory Capability Monitoring System (EULabCap)—Report on 2016 Survey of EU/EEA Country Capabilities and Capacities. 2018. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/2016_EULabCap_EUreport_web_300418_final.pdf (accessed on 11 October 2021).
- European Centre for Disease Prevention and Control; European Food Safety Authority. Multi-Country Outbreak of Salmonella Enteritidis Infections Linked to Eggs, Third Update–6 February 2020. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/salmonella-enteritidis-rapid-outbreak-assessment-third-update.pdf (accessed on 11 October 2021).
- Michael, G.; Schwarz, S. Antimicrobial resistance in zoonotic nontyphoidal Salmonella: An alarming trend? Clin. Microbiol. Infect. 2016, 22, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority; European Centre for Disease Prevention and Control. The European Union Summary Report on Antimicrobial Resistance in zoonotic and indicator bacteria from humans, animals and food in 2017/2018. EFSA J. 2020, 18, e06007. [Google Scholar] [CrossRef] [Green Version]
- Imre, K.; Herman, V.; Morar, A. Scientific Achievements in the study of the occurrence and antimicrobial susceptibility profile of major foodborne pathogenic bacteria in foods and food processing environments in Romania: Review of the last decade. BioMed Res. Int. 2020, 2020, 5134764. [Google Scholar] [CrossRef] [PubMed]
- Magnet, S.; Blanchard, J.S. Molecular insights into aminoglycoside action and resistance. Chem. Rev. 2005, 105, 477–498. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Biological Hazards (EFSA BIOHAZ Panel); Koutsoumanis, K.; Allende, A.; Alvarez-Ordóñez, A.; Bolton, D.; Bover-Cid, S.; Chemaly, M.; Davies, R.; De Cesare, A.; Hilbert, F.; et al. Whole genome sequencing and metagenomics for outbreak investigation, source attribution and risk assessment of food-borne microorganisms. EFSA J. 2019, 17, e05898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achtman, M.; Wain, J.; Weill, F.-X.; Nair, S.; Zhou, Z.; Sangal, V.; Krauland, M.; Hale, J.L.; Harbottle, H.; Uesbeck, A.; et al. Multilocus Sequence Typing as a Replacement for Serotyping in Salmonella enterica. PLoS Pathog. 2012, 8, e1002776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, M.E.; Alikhan, N.-F.; Dallman, T.J.; Zhou, Z.; Grant, K.; Maiden, M.C. Comparative analysis of core genome MLST and SNP typing within a European Salmonella serovar Enteritidis outbreak. Int. J. Food Microbiol. 2018, 274, 1–11. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control. Laboratory Standard Operating Procedure for Multiple-Locus Variable-Number Tandem Repeat Analysis of Salmonella Enterica Serotype Enteritidis. 2016. Available online: https://www.ecdc.europa.eu/sites/default/files/media/en/publications/Publications/Salmonella-Enteritidis-Laboratory-standard-operating-procedure.pdf (accessed on 11 October 2021).
- Hopkins, K.; Peters, T.M.; de Pinna, E.; Wain, J. Standardisation of multilocus variable-number tandem-repeat analysis (MLVA) for subtyping of Salmonella enterica serovar Enteritidis. Eurosurveillance 2011, 16, 19942. [Google Scholar] [CrossRef]
- Alikhan, N.-F.; Zhou, Z.; Sergeant, M.J.; Achtman, M. A genomic overview of the population structure of Salmonella. PLoS Genet. 2018, 14, e1007261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaas, R.S.; Leekitcharoenphon, P.; Aarestrup, F.; Lund, O. Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS ONE 2014, 9, e104984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Allesøe, R.; Joensen, K.G.; Cavaco, L.M.; Lund, O.; Aarestrup, F.M. PointFinder: A novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother. 2017, 72, 2764–2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coipan, C.E.; Dallman, T.J.; Brown, D.; Hartman, H.; Van Der Voort, M.; Berg, R.R.V.D.; Palm, D.; Kotila, S.; Van Wijk, T.; Franz, E. Concordance of SNP- and allele-based typing workflows in the context of a large-scale international Salmonella Enteritidis outbreak investigation. Microb. Genom. 2020, 6, e000318. [Google Scholar] [CrossRef] [PubMed]
- Di Marcantonio, L.; Janowicz, A.; Zilli, K.; Romantini, R.; Bilei, S.; Paganico, D.; Persiani, T.; Di Donato, G.; Di Giannatale, E. Genomic comparison of salmonella enteritidis strains isolated from laying hens and humans in the abruzzi region during 2018. Pathogens 2020, 9, 349. [Google Scholar] [CrossRef] [PubMed]
- The European Committee on Antimicrobial Susceptibility Testing. Clinical Breakpoint Tables 9.0. 2019. Available online: www.eucast.org (accessed on 11 October 2021).
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing, 28th ed.; CLSI supplement M100; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018. [Google Scholar]
- Hunter, P.R.; Gaston, M.A. Numerical index of the discriminatory ability of typing systems: An application of Simpson’s index of diversity. J. Clin. Microbiol. 1988, 26, 2465–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundmann, H.; Hori, S.; Tanner, G. Determining confidence intervals when measuring genetic diversity and the discriminatory abilities of typing methods for microorganisms. J. Clin. Microbiol. 2001, 39, 4190–4192. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| MLVA Genotype/ Cluster | Allelic Profile | No. of Strains | County Distribution 1 | Phenotypic Antibiotic Susceptibility Profiles 2(No. of Strains) | ||||
|---|---|---|---|---|---|---|---|---|
| 2016 | 2017 | 2018 | 2019 | 2020 | ||||
| G1/CI | 2-11-7-3-2 | 120 | BZ, CV, DB, IS *,3 | AB, BR, BT, BZ, CL, CT, CV, DB, HR *, IS *, OT, VS | B *, BR, BT, BV, BZ, DB, HR *, IS *, MH, OT, TL, VN | B, BT, BZ, DB, MH, MM *, OT, TM | AB * | Susceptible (83) S3 R (1) NA RCIP RPEF R (35) NA RCIPRPEF RS3 R (1) |
| G2/CI | 2-10-7-3-2 | 30 | IS, VN | BT, CV | BZ, MH, MM *, SV | B, BR, BZ, IS, MH, MM, TM | Susceptible (29) S3 R (1) | |
| G3/CI | 2-12-7-3-2 | 27 | BN, BR, BZ, CV, DB, IS * | B, BV, BZ, DB, PH * | B, BZ, DB | Susceptible (4) NA RCIP RPEF R (23) | ||
| G4/CII | 2-10-12-5-1 | 14 | VS∗ | Susceptible (14) | ||||
| G5/CI | 2-11-8-3-2 | 9 | IS | CV, DB | BZ, DB | BR, DB | Susceptible (6) NA RCIP RPEF R (3) | |
| G6/CI | 2-9-7-3-2 | 9 | IS | BZ, CV, DB | BZ, CV, SM | DB | Susceptible (8) AMP R (1) | |
| G7 | 3-10-5-4-1 | 5 | DB, MH | MM * | Susceptible (5) | |||
| G8/CI | 2-11-6-3-2 | 4 | DB * | CV, DB | Susceptible (2) S R (1) AMP RNA RCIP RPEF R (1) | |||
| G9/CI | 2-13-7-3-2 | 4 | IS | Susceptible (4) | ||||
| G10/CI | 2-10-6-3-2 | 2 | BR, DB | Susceptible (2) | ||||
| G11/CI | 2-12-9-3-2 | 2 | BZ | B | Susceptible (1) NA RCIP RPEF R (1) | |||
| G12/CIII | 3-11-5-3-1 | 2 | DB | CL | Susceptible (1) NA RCIP RPEF R (1) | |||
| G13/CIII | 3-11-5-4-1 | 2 | BT | BT | Susceptible (2) | |||
| G14/CIII | 3-9-5-4-1 | 2 | MH | MM | Susceptible (1) NA RCIP RPEF R (1) | |||
| G15/CI | 2-10-4-3-2 | 1 | B | NA R (1) | ||||
| G16/CI | 2-10-8-3-2 | 1 | SV | Susceptible (1) | ||||
| G17/CI | 2-10-8-5-2 | 1 | DB | Susceptible (1) | ||||
| G18 | 2-10-9-6-1 | 1 | DB | Susceptible (1) | ||||
| G19/CII | 2-11-12-5-1 | 1 | VS | Susceptible (1) | ||||
| G20/CII | 2-11-14-5-1 | 1 | B | Susceptible (1) | ||||
| G21/CI | 2-11-7-4-2 | 1 | DB | Susceptible (1) | ||||
| G22/CI | 2-11-8-5-2 | 1 | B | Susceptible (1) | ||||
| G23 | 2-12-6-3-2 | 1 | CV | Susceptible (1) | ||||
| G24/CII | 2-14-11-5-1 | 1 | DB | NA R (1) | ||||
| G25 | 2-15-11-4-1 | 1 | B | Susceptible (1) | ||||
| G26/CI | 2-9-8-3-2 | 1 | BV | Susceptible (1) | ||||
| G27/CI | 2-9-8-4-2 | 1 | BT | Susceptible (1) | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usein, C.-R.; Oprea, M.; Ciontea, A.S.; Dinu, S.; Cristea, D.; Zota, L.C.; Kotila, S. A Snapshot of the Genetic Diversity of Salmonella Enteritidis Population Involved in Human Infections in Romania Taken in the European Epidemiological Context. Pathogens 2021, 10, 1490. https://doi.org/10.3390/pathogens10111490
Usein C-R, Oprea M, Ciontea AS, Dinu S, Cristea D, Zota LC, Kotila S. A Snapshot of the Genetic Diversity of Salmonella Enteritidis Population Involved in Human Infections in Romania Taken in the European Epidemiological Context. Pathogens. 2021; 10(11):1490. https://doi.org/10.3390/pathogens10111490
Chicago/Turabian StyleUsein, Codruta-Romanita, Mihaela Oprea, Adriana Simona Ciontea, Sorin Dinu, Daniela Cristea, Lavinia Cipriana Zota, and Saara Kotila. 2021. "A Snapshot of the Genetic Diversity of Salmonella Enteritidis Population Involved in Human Infections in Romania Taken in the European Epidemiological Context" Pathogens 10, no. 11: 1490. https://doi.org/10.3390/pathogens10111490