
Human Polyomaviruses (HPyV) in Wastewater and Environmental Samples from the Lisbon Metropolitan Area: Detection and Genetic Characterization of Viral Structural Protein-Coding Sequences

 , ,
, ,  , and
, and
Abstract
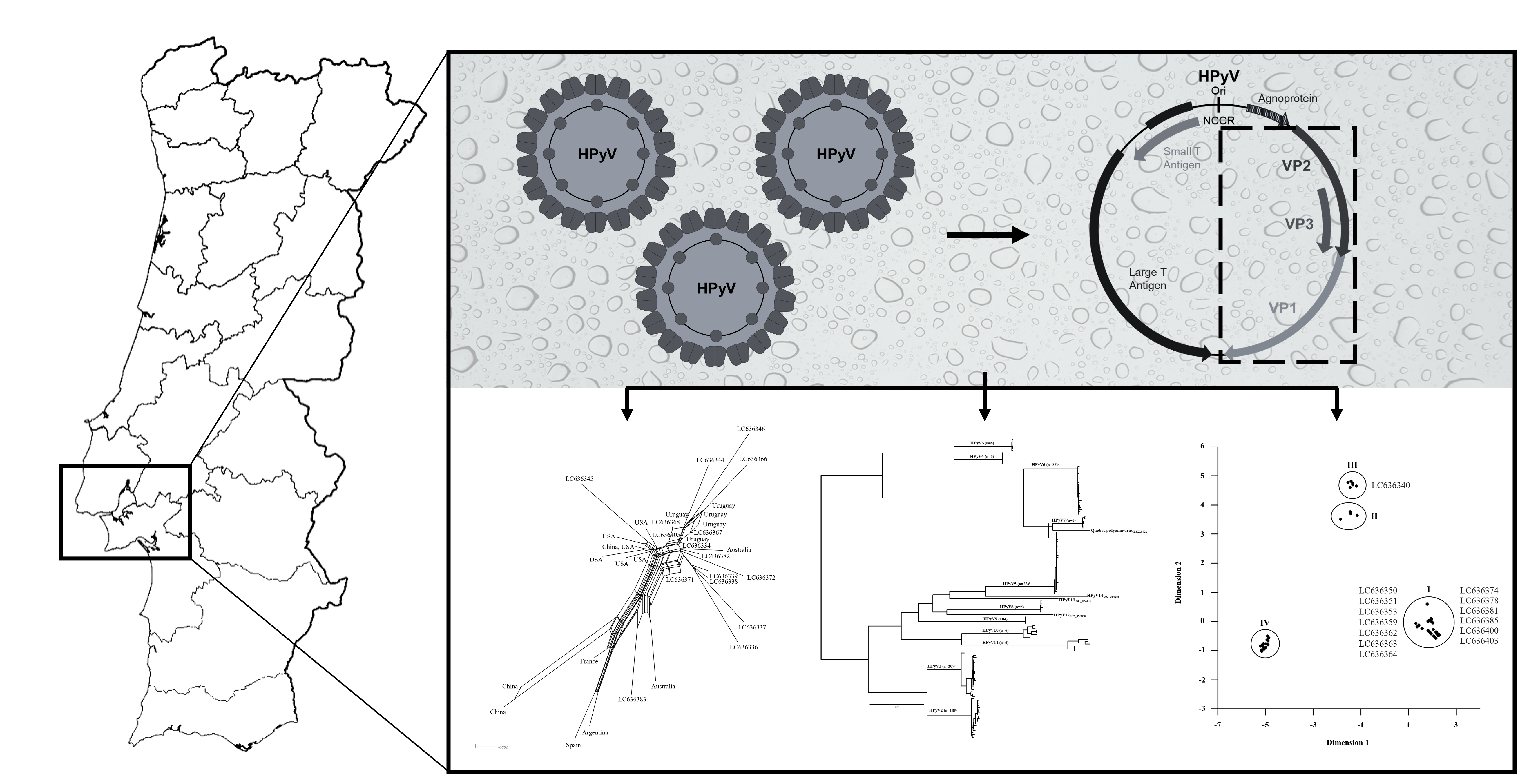
:
1. Introduction
2. Results
2.1. Primer Design and Touch-Down PCR Setup
2.2. Analysis of PCR Amplification Results
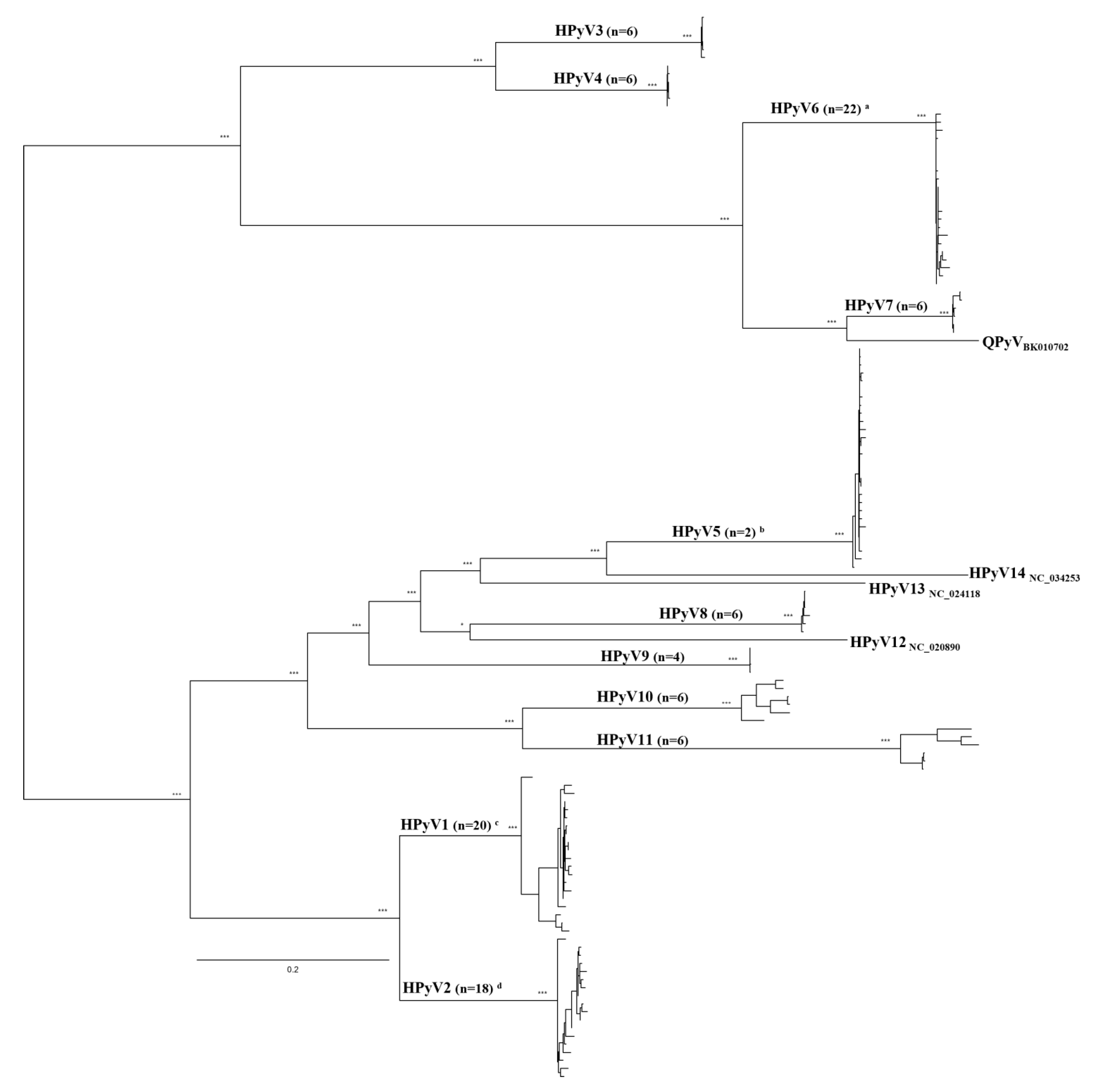
2.3. Genetic Characterization of Viral Sequences
3. Discussion
4. Materials and Methods
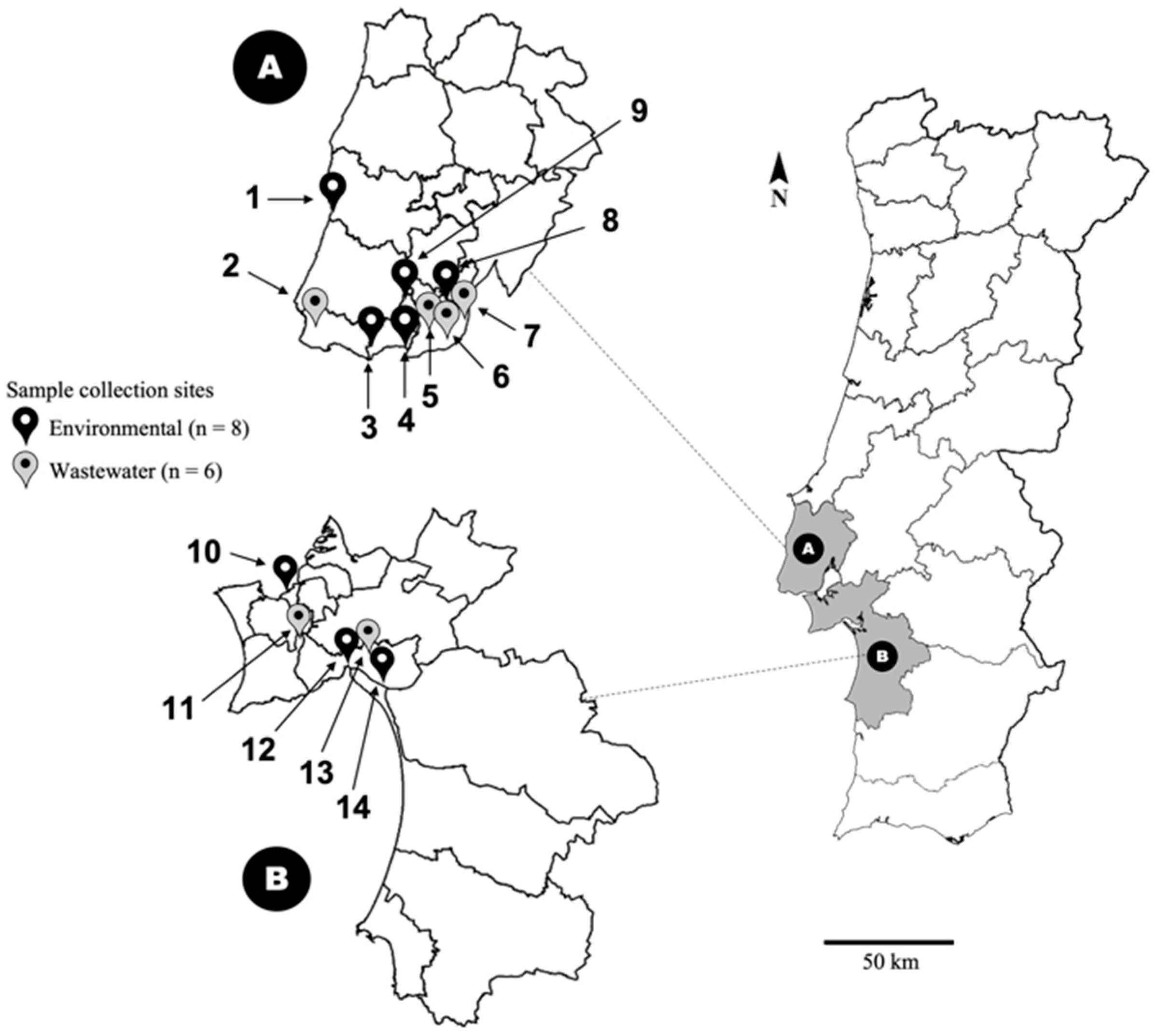
4.1. Virus Concentration by Skimmed-Milk Flocculation and DNA Extraction
4.2. Primer Design and Touch-Down Multiplex-PCR Set-Up
4.3. DNA Cloning and Sequencing
4.4. Nucleotide Sequence Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johne, R.; Buck, C.B.; Allander, T.; Atwood, W.J.; Garcea, R.L.; Imperiale, M.J.; Major, E.O.; Ramqvist, T.; Norkin, L.C. Taxonomical developments in the family Polyomaviridae. Arch. Virol. 2011, 156, 1627–1634. [Google Scholar] [CrossRef] [Green Version]
- Moens, U.; Calvignac-Spencer, S.; Lauber, C.; Ramqvist, T.; Feltkamp, M.C.; Daugherty, M.D.; Verschoor, E.J.; Ehlers, B. Report on the Taxonomy of the Polyomaviridae. Virus Taxonomy: 2020 Release. 2020. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5656788/ (accessed on 31 April 2020).
- Calvignac-Spencer, S.; Feltkamp, M.C.W.; Daugherty, M.D.; Moens, U.; Ramqvist, T.; Johne, R.; Ehlers, B. A taxonomy update for the family Polyomaviridae. Arch. Virol. 2016, 161, 1739–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kean, J.M.; Rao, S.; Wang, M.; Garcea, R.L. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009, 5, e1000363. [Google Scholar] [CrossRef] [Green Version]
- Van Der Meijden, E.; Bialasiewicz, S.; Rockett, R.J.; Tozer, S.J.; Sloots, T.P.; Feltkamp, M.C.W. Different serologic behavior of MCPyV, TSPyV, HPyV6, HPyV7 and HPyV9 polyomaviruses found on the skin. PLoS ONE 2013, 8, e81078. [Google Scholar] [CrossRef] [Green Version]
- Rinaldo, C.H.; Tylden, G.D.; Sharma, B.N. The human polyomavirus BK (BKPyV): Virological background and clinical implications. Apmis 2013, 121, 728–745. [Google Scholar] [CrossRef]
- Wiedinger, K.; Bitsaktsis, C.; Chang, S. Reactivation of human polyomaviruses in immunocompromised states. J. Neurovirol. 2014, 20, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalianis, T.; Hirsch, H.H. Human polyomaviruses in disease and cancer. Virology 2013, 437, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, S.D.; Field, A.M.; Coleman, D.V. New human papovavirus (B.K.) isolated from urine after renal transplantation. Lancet 1970, 297, 1253–1257. [Google Scholar] [CrossRef]
- Fiksdal, L.; Maki, J.S.; LaCroix, S.J.; Staley, J.T. Survival and detection of Bacteroides spp., prospective indicator bacteria. Appl. Environ. Microbiol. 1985, 49, 148–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harwood, V.J.; Levine, A.D.; Scott, T.M.; Chivukula, V.; Lukasik, J.; Farrah, S.R.; Rose, J.B. Validity of the indicator organism paradigm for pathogen reduction in reclaimed water and public health protection. Appl. Environ. Microbiol. 2005, 71, 3163–3170. [Google Scholar] [CrossRef] [Green Version]
- McQuaig, S.; Griffith, J.; Harwood, V.J. Association of fecal indicator bacteria with human viruses and microbial source tracking markers at coastal beaches impacted by nonpoint source pollution. Appl. Environ. Microbiol. 2012, 78, 6423–6432. [Google Scholar] [CrossRef] [Green Version]
- Rachmadi, A.T.; Torrey, J.R.; Kitajima, M. Human polyomavirus: Advantages and limitations as a human-specific viral marker in aquatic environments. Water Res. 2016, 105, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Torres, C. Evolution and molecular epidemiology of polyomaviruses. Infect. Genet. Evol. 2020, 79, 104150. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Starrett, G.J.; Sappington, A.; Kostic, A.; Koren, S.; Buck, C.B.; Phillippy, A.M. Mash Screen: High-throughput sequence containment estimation for genome discovery. Genome Biol. 2019, 20, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korup, S.; Rietscher, J.; Calvignac-Spencer, S.; Trusch, F.; Hofmann, J.; Moens, U.; Sauer, I.; Voigt, S.; Schmuck, R.; Ehlers, B. Identification of a novel human polyomavirus in organs of the gastrointestinal tract. PLoS ONE 2013, 8, e58021. [Google Scholar] [CrossRef] [Green Version]
- Fahsbender, E.; Altan, E.; Estrada, M.; Seguin, M.A.; Young, P.; Leutenegger, C.M.; Delwart, E. Lyon-IARC polyomavirus DNA in feces of diarrheic cats. Microbiol. Resour. Announc. 2019, 8, 54–56. [Google Scholar] [CrossRef] [Green Version]
- Prezioso, C.; Van Ghelue, M.; Pietropaolo, V.; Moens, U. Detection of quebec polyomavirus DNA in samples from different patient groups. Microorganisms 2021, 9, 1082. [Google Scholar] [CrossRef]
- Korbie, D.J.; Mattick, J.S. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nat. Protoc. 2008, 3, 1452–1456. [Google Scholar] [CrossRef]
- Martel-Jantin, C.; Filippone, C.; Tortevoye, P.; Afonso, P.V.; Betsem, E.; Descorps-Declere, S.; Nicol, J.T.J.; Touzé, A.; Coursaget, P.; Crouzat, M.; et al. Molecular epidemiology of merkel cell polyomavirus: Evidence for geographically related variant genotypes. J. Clin. Microbiol. 2014, 52, 1687–1690. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Gibson, P.E.; Booth, J.C.; Clewley, J.P. Genomic typing of BK virus in clinical specimens by direct sequencing of polymerase chain reaction products. J. Med. Virol. 1993, 41, 11–17. [Google Scholar] [CrossRef]
- Morel, V.; Martin, E.; François, C.; Helle, F.; Faucher, J.; Mourez, T.; Choukroun, G.; Duverlie, G.; Castelain, S.; Brochot, E. A simple and reliable strategy for BK virus subtyping and subgrouping. J. Clin. Microbiol. 2017, 55, 1177–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auit, G.S.; Stoner, G.L. Two major types of JC virus defined in progressive multifocal leukoencephalopathy brain by early and late coding region DNA sequences. J. Gen. Virol. 1992, 2669–2678. [Google Scholar]
- Agostini, H.T.; Yanagihara, R.; Davis, V.; Ryschkewitsch, C.F.; Stoner, G.L. Asian genotypes of JC virus in Native Americans and in a Pacific Island population: Markers of viral evolution and human migration. Proc. Natl. Acad. Sci. USA 1997, 94, 14542–14546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baez, C.F.; Diaz, N.C.; Venceslau, M.T.; Luz, F.B.; Guimarães, M.A.A.M.; Zalis, M.G.; Varella, R.B. Phylogenetic and structural analysis of merkel cell polyomavirus VP1 in Brazilian samples. Virus Res. 2016, 221, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Iaconelli, M.; Petricca, S.; Libera, S.D.; Di Bonito, P.; La Rosa, G. First detection of human papillomaviruses and human polyomaviruses in river waters in Italy. Food Environ. Virol. 2015, 7, 309–315. [Google Scholar] [CrossRef]
- Kokkinos, P.; Ziros, P.; Meri, D.; Filippidou, S.; Kolla, S.; Galanis, A.; Vantarakis, A. Environmental surveillance. An additional/alternative approach for virological surveillance in Greece? Int. J. Environ. Res. Public Health 2011, 8, 1914–1922. [Google Scholar] [CrossRef] [Green Version]
- Kirs, M.; Caffaro-filho, R.A.; Wong, M.; Harwood, V.J.; Moravcik, P.; Fujioka, S. Human-associated Bacteroides spp. and human polyomaviruses as microbial source tracking markers in Hawaii. Appl. Environ. Microbiol. 2017, 82, 6757–6767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calgua, B.; Fumian, T.; Rusiñol, M.; Rodriguez-Manzano, J.; Mbayed, V.A.; Bofill-Mas, S.; Miagostovich, M.; Girones, R. Detection and quantification of classic and emerging viruses by skimmed-milk flocculation and PCR in river water from two geographical areas. Water Res. 2013, 47, 2797–2810. [Google Scholar] [CrossRef] [Green Version]
- Fumian, T.M.; Guimarães, F.R.; Vaz, B.J.P.; Da Silva, M.T.T.; Muylaert, F.F.; Bofill-Mas, S.; Gironés, R.; Leite, J.P.G.; Miagostovich, M.P. Molecular detection, quantification and characterization of human polyomavirus JC from waste water in Rio de Janeiro, Brazil. J. Water Health 2010, 8, 438–445. [Google Scholar] [CrossRef]
- Rusiñol, M.; Fernandez-Cassi, X.; Hundesa, A.; Vieira, C.; Kern, A.; Eriksson, I.; Ziros, P.; Kay, D.; Miagostovich, M.; Vargha, M.; et al. Application of human and animal viral microbial source tracking tools in fresh and marine waters from five different geographical areas. Water Res. 2014, 59, 119–129. [Google Scholar] [CrossRef] [Green Version]
- McQuaig, S.M.; Scott, T.M.; Lukasik, J.O.; Paul, J.H.; Harwood, V.J. Quantification of human polyomaviruses JC virus and BK Virus by TaqMan quantitative PCR and comparison to other water quality indicators in water and fecal samples. Appl. Environ. Microbiol. 2009, 75, 3379–3388. [Google Scholar] [CrossRef] [Green Version]
- Torres, C.; Barrios, M.E.; Cammarata, R.V.; Cisterna, D.M.; Estrada, T.; Martini Novas, S.; Cahn, P.; Blanco Fernández, M.D.; Mbayed, V.A. High diversity of human polyomaviruses in environmental and clinical samples in Argentina: Detection of JC, BK, Merkel-cell, Malawi, and human 6 and 7 polyomaviruses. Sci. Total Environ. 2016, 542, 192–202. [Google Scholar] [CrossRef]
- Harwood, V.J.; Staley, C.; Badgley, B.D.; Borges, K.; Korajkic, A. Microbial source tracking markers for detection of fecal contamination in environmental waters: Relationships between pathogens and human health outcomes. FEMS Microbiol. Rev. 2014, 38, 1–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochelle-Newall, E.; Nguyen, T.M.H.; Le, T.P.Q.; Sengtaheuanghoung, O.; Ribolzi, O. A short review of fecal indicator bacteria in tropical aquatic ecosystems: Knowledge gaps and future directions. Front. Microbiol. 2015, 6, 308. [Google Scholar] [CrossRef] [PubMed]
- McQuaig, S.M.; Scott, T.M.; Harwood, V.J.; Farrah, S.R.; Lukasik, J.O. Detection of human-derived fecal pollution in environmental waters by use of a PCR-based human polyomavirus assay. Appl. Environ. Microbiol. 2006, 72, 7567–7574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bofill-Mas, S.; Pina, S.; Girones, R. Documenting the epidemiologic patterns of polyomaviruses in human populations by studying their presence in urban sewage. Appl. Environ. Microbiol. 2000, 66, 238–245. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, W.; Wan, C.; Goonetilleke, A.; Gardner, T. Evaluating sewage-associated JCV and BKV polyomaviruses for sourcing human fecal pollution in a coastal river in Southeast Queensland, Australia. J. Environ. Qual. 2010, 39, 1743–1750. [Google Scholar] [CrossRef] [Green Version]
- Jurzik, L.; Hamza, I.A.; Wilhelm, M. Investigating the Reduction of Human Adenovirus (HAdV) and Human Polyomavirus (HPyV) in a Sewage Treatment Plant with a Polishing Pond as a Tertiary Treatment. Water. Air. Soil Pollut. 2015, 226, 284. [Google Scholar] [CrossRef]
- Hewitt, J.; Greening, G.E.; Leonard, M.; Lewis, G.D. Evaluation of human adenovirus and human polyomavirus as indicators of human sewage contamination in the aquatic environment. Water Res. 2013, 47, 6750–6761. [Google Scholar] [CrossRef] [PubMed]
- Bofill-Mas, S.; Albinana-Gimenez, N.; Clemente-Casares, P.; Hundesa, A.; Rodriguez-Manzano, J.; Allard, A.; Calvo, M.; Girones, R. Quantification and stability of human adenoviruses and polyomavirus JCPyV in wastewater matrices. Appl. Environ. Microbiol. 2006, 72, 7894–7896. [Google Scholar] [CrossRef] [Green Version]
- Cantalupo, P.G.; Calgua, B.; Zhao, G.; Hundesa, A.; Wier, A.D.; Katz, J.P.; Grabe, M.; Hendrix, R.W.; Girones, R.; Wang, D.; et al. Raw sewage harbors diverse viral populations. MBio 2011, 2, e00180-11. [Google Scholar] [CrossRef] [Green Version]
- Torres, C.; Barrios, M.E.; Cammarata, R.V.; Victoria, M.; Fernandez-Cassi, X.; Bofill-Mas, S.; Colina, R.; Blanco Fernández, M.D.; Mbayed, V.A. Phylodynamics of Merkel-cell polyomavirus and human polyomavirus 6: A long-term history with humans. Mol. Phylogenet. Evol. 2018, 126, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Rockett, R.J.; Sloots, T.P.; Bowes, S.; O’Neill, N.; Ye, S.; Robson, J.; Whiley, D.M.; Lambert, S.B.; Wang, D.; Nissen, M.D.; et al. Detection of Novel Polyomaviruses, TSPyV, HPyV6, HPyV7, HPyV9 and MWPyV in Feces, Urine, Blood, Respiratory Swabs and Cerebrospinal Fluid. PLoS ONE 2013, 8, e62764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bofill-Mas, S.; Rodriguez-Manzano, J.; Calgua, B.; Carratala, A.; Girones, R. Newly described human polyomaviruses Merkel Cell, KI and WU are present in urban sewage and may represent potential environmental contaminants. Virol. J. 2010, 7, 141. [Google Scholar] [CrossRef] [Green Version]
- Babakir-Mina, M.; Ciccozzi, M.; Alteri, C.; Polchi, P.; Picardi, A.; Greco, F.; Lucarelli, G.; Arcese, W.; Perno, C.F.; Perno, M. Excretion of the novel polyomaviruses KI and WU in the stool of patients with hematological disorders. Antivir. Ther. 2006, 55, 52–55. [Google Scholar] [CrossRef]
- Siebrasse, E.A.; Bauer, I.; Holtz, L.R.; Le, B.; Lassa-claxton, S.; Canter, C.; Hmiel, P.; Shenoy, S.; Sweet, S.; Turmelle, Y.; et al. Human polyomaviruses in children undergoing transplantation, United States, 2008–2010. Emerg. Infect. Dis. 2012, 18, 1676–1679. [Google Scholar] [CrossRef]
- Pinheiro, M.S.; Mendes, G.S.; Santos, N. Human polyomaviruses 10 and 11 in faecal samples from Brazilian children. Braz. J. Microbiol. 2020, 51, 585–591. [Google Scholar] [CrossRef]
- Zheng, H.Y.; Nishimoto, Y.; Chen, Q.; Hasegawa, M.; Zhong, S.; Ikegaya, H.; Ohno, N.; Sugimoto, C.; Takasaka, T.; Kitamura, T.; et al. Relationships between BK virus lineages and human populations. Microbes Infect. 2007, 9, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, Y.; Zheng, H.Y.; Zhong, S.; Ikegaya, H.; Chen, Q.; Sugimoto, C.; Kitamura, T.; Yogo, Y. An Asian origin for subtype IV BK virus based on phylogenetic analysis. J. Mol. Evol. 2007, 65, 103–111. [Google Scholar] [CrossRef]
- Sugimoto, C.; Kitamura, T.; Guo, J.; Al-ahdal, M.N.; Shchelkunov, S.N.; Otova, B.; Ondrejka, P.; Chollet, J.-Y.; El-Safi, S.; Ettayebi, M.; et al. Typing of urinary JC virus DNA offers a novel means of tracing human migrations. Proc. Natl. Acad. Sci. USA 1997, 94, 9191–9196. [Google Scholar] [CrossRef] [Green Version]
- Yogo, Y.; Sugimoto, C.; Zheng, H.Y.; Ikegaya, H.; Takasaka, T.; Kitamura, T. JC virus genotyping offers a new paradigm in the study of human populations. Rev. Med. Virol. 2004, 14, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Stoner, G.L.; Jobes, D.V.; Fernandez, M.; Agostini, H.T.; Chima, S.C.; Ryschkewitsch, C.F. JC virus as a marker of human migration to the Americas. Microbes Infect. 2000, 2, 1905–1911. [Google Scholar] [CrossRef]
- Saruwatari, L.; Sugimoto, C.; Kitamura, T.; Ohno, N.; Sakai, E.; Shresta, P.; Hoa, B.K.; Phi, P.T.P.; An, H.P.H.; Tuyet, N.T.A.; et al. Asian domains of four major genotypes of JC virus, Af2, B1-b, CY and SC. Arch. Virol. 2002, 147, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Agostini, T.; Shishido-hara, Y.; Baumhefner, R.W.; Singer, E.J.; Ryschkewitsch, C.F.; Stoner, G.L. JC virus Type 2: Definition of subtypes based on DNA sequence analysis of ten complete genomes. J. Gen. Virol. 1998, 79, 1143–1151. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Wang, J.C.; Deckhut, A.; Joseph, B.C.; Eberwein, P.; Cubitt, C.L.; Ryschkewitsch, C.F.; Agostini, H.T.; Stoner, G.L. Chinese Strains ( Type 7 ) of JC Virus Are Afro-Asiatic in Origin But Are Phylogenetically Distinct from the Mongolian and Indian Strains ( Type 2D ) and the Korean and Japanese Strains (Type 2A)*. J. Mol. Evol. 2004, 58, 568–583. [Google Scholar] [CrossRef] [PubMed]
- Jobes, D.V.; Friedlaender, J.S.; Mgone, C.S.; Agostini, H.T.; Koki, G. New JC virus (JCV) genotypes from Papua New Guinea and Micronesia (Type 8 and Type 2E) and evolutionary analysis of 32 complete JCV genomes. Arch. Virol. 2001, 146, 2097–2113. [Google Scholar] [CrossRef] [PubMed]
- Takasaka, T.; Kitamura, T.; Sugimoto, C.; Guo, J.; Zheng, H.Y.; Yogo, Y. Phylogenetic analysis of major African genotype (Af2) of JC virus: Implications for origin and dispersals of modern Africans. Am. J. Phys. Anthropol. 2006, 129, 465–472. [Google Scholar] [CrossRef]
- Agostini, H.T.; Deckhut, A.; Jobes, D.V.; Girones, R.; Schlunck, G.; Prost, M.G.; Frias, C.; Pérez-Trallero, E.; Ryschkewitsch, C.F.; Stoner, G.L. Genotypes of JC virus in East, Central and Southwest Europe. J. Gen. Virol. 2001, 82, 1221–1331. [Google Scholar] [CrossRef] [Green Version]
- Pagani, E.; Delbue, S.; Mancuso, R.; Borghi, E.; Tarantini, L.; Ferrante, P. Molecular analysis of JC virus genotypes circulating among the Italian healthy population. J. Neurovirol. 2003, 9, 559–566. [Google Scholar] [CrossRef]
- Hjelmsø, M.H.; Hellmér, M.; Fernandez-Cassi, X.; Timoneda, N.; Lukjancenko, O.; Seidel, M.; Elsässer, D.; Aarestrup, F.M.; Löfström, C.; Bofill-Mas, S.; et al. Evaluation of methods for the concentration and extraction of viruses from sewage in the context of metagenomic sequencing. PLoS ONE 2017, 12, e0170199. [Google Scholar] [CrossRef] [Green Version]
- Calgua, B.; Mengewein, A.; Grunert, A.; Bofill-Mas, S.; Clemente-Casares, P.; Hundesa, A.; Wyn-Jones, A.P.; López-Pila, J.M.; Girones, R. Development and application of a one-step low cost procedure to concentrate viruses from seawater samples. J. Virol. Methods 2008, 153, 79–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, H.; Leitner, T. PrimerDesign-M: A multiple-alignment based multiple-primer design tool for walking across variable genomes. Bioinformatics 2015, 31, 1472–1474. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.T.; Niemela, S.L.; Miller, R.H. One-step preparation of competent Escherichia coli: Transformation and storage of bacterial cells in the same solution. Proc. Natl. Acad. Sci. USA 1989, 86, 2172–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.M.; et al. Genome Detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Strimmer, K.; von Haeseler, A. Likelihood-mapping: A simple method to visualize phylogenetic content of a sequence alignment. Proc. Natl. Acad. Sci. USA 1997, 94, 6815–6819. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.A.; Strimmer, K.; Vingron, M.; von Haeseler, A. TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 2002, 18, 502–504. [Google Scholar] [CrossRef] [Green Version]
- Trifinopoulos, J.; Nguyen, L.; Von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, 232–235. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, S.Y.W.; Phillips, M.J.; Drummond, A.J.; Cooper, A. Accuracy of rate estimation using relaxed-clock models with a critical focus on the early metazoan radiation. Mol. Biol. Evol. 2005, 22, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Rambaut, a.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Reaction | Primers (5′-3′) | Number of Mismatches a | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HPyV1 | HPyV2 | HPyV3 | HPyV4 | HPyV5 | HPyV6 | HPyV7 | HPyV8 | HPyV9 | HPyV10 | HPyV11 | |||
| PCR-A | 1st round | HPyV1/2Fo: CTGCTCCTCAATGGATGTTGC (from 1489 to 1509) b | 0 | 0 | - | - | 10 | - | - | 7 | 9 | 7 | 17 |
| HPyV1/2Ro: ATCATRTCTGGGTCCCCTGGAAG (from 2606 to 2584) b | 0 | 0 | - | - | 8 | - | - | 4 | 7 | 7 | 8 | ||
| HPyV5Fo: GAAAATAGCTTGCTGCATTCTG (from 879 to 900) c | 35 | 15 | - | - | 0 | - | - | 7 | 10 | 10 | 14 | ||
| HPyV5Ro: GGGCCCACTCCATTCTCATC (from 1979 to 1960) c | 5 | 6 | - | - | 0 | - | - | 2 | 2 | 5 | 8 | ||
| HPyV8/9Fo: AGGAGGRGCAMATCAAAGAG (from 1198 to 1217) d | 3 | 2 | - | - | 9 | - | - | 0 | 0 | 3 | 10 | ||
| HPyV8/9Ro: ATAAAYTCTGACTTCTTCMAC (from 2336 to 2316) d | 6 | 5 | - | - | 4 | - | - | 0 | 0 | 10 | 8 | ||
| HPyV10/11Fo: CCTGGATAYAGACAMTTTSA (from 806 to 825) e | 9 | 17 | - | - | 10 | - | - | 11 | 13 | 0 | 0 | ||
| HPyV10/11Ro: TTAMAGGATAAGGATTTCTVA (from 2334 to 2313) e | 7 | 8 | - | - | 6 | - | - | 5 | 1 | 0 | 1 | ||
| Expected amplicon size (bp) f | 1113 | 1094 | - | - | 1101 | - | - | 1139 | 1136 | 1529 | 1474 | ||
| PCR-A | 2nd round | HPyV1/2Fi: GTACGGGACTGTAACACCTGC (from 1526 to 1546) b | 0 | 0 | - | - | 12 | - | - | 8 | 8 | 11 | 14 |
| HPyV1/2Ri: CCATACATAGGCTGCCCATC (from 2534 to 2515) b | 0 | 0 | - | - | 7 | - | - | 7 | 7 | 3 | 7 | ||
| HPyV5Fi: CAATCAAACCTAGTGAATCTG (from 951 to 971) c | 31 | 18 | - | - | 0 | - | - | 12 | 10 | 13 | 10 | ||
| HPyV5Ri: GGATCAGGACACCATACTTC (from 1859 to 1840) c | 7 | 5 | - | - | 0 | - | - | 7 | 5 | 6 | 4 | ||
| HPyV8/9Fi: GGWTTGTATGGTGATATAAC (from 1250 to1269) d | 7 | 6 | - | - | 9 | - | - | 0 | 0 | 5 | 7 | ||
| HPyV8/9Ri: ATTAAARTAYCTAGGTAGGCCTCT (from 2192 to 2170) d | 4 | 5 | - | - | 6 | - | - | 0 | 0 | 5 | 5 | ||
| HPyV10/11Fi: AGAGCTTTTTGGGARGCTKT (from 881 to 900) e | 8 | 7 | - | - | 14 | - | - | 6 | 10 | 0 | 1 | ||
| HPyV10/11Ri: CCCAGGCCTCYACWGGATAR (from 2055 to 2036) e | 3 | 6 | - | - | 5 | - | - | 4 | 5 | 0 | 1 | ||
| Expected amplicon size (bp) f | 989 | 985 | - | - | 909 | - | - | 943 | 940 | 1175 | 1136 | ||
| PCR-B | 1st round | HPyV3/4Fo: GGACGTGTTCAATAGAATTGC (from 980 to 1000) g | - | - | 0 | 0 | - | 10 | 8 | - | - | - | - |
| HPyV3/4Ro: CCAATGCCATTTTCATCCAA (from 2282 to 2263) g | - | - | 0 | 0 | - | 5 | 4 | - | - | - | - | ||
| HPyV6/7Fo: GACTCGGCCCAAGARTTGG (from 708 to 726) h | - | - | 9 | 13 | - | 0 | 0 | - | - | - | - | ||
| HPyV6/7Ro: GCACCTGTGGCTTCTGRGG (from 2220 to 2202) h | - | - | 7 | 6 | - | 0 | 0 | - | - | - | - | ||
| Expected amplicon size (bp) f | - | - | 1303 | 1318 | - | 1513 | 1513 | - | - | - | - | ||
| PCR-B | 2nd round | HPyV3/4Fi: CATCATATTACAATRCGGGG (from 1015 to 1034) g | - | - | 0 | 0 | - | 8 | 7 | - | - | - | - |
| HPyV3/4Ri: GTTTCCATTCTRTACAGCTC (from 1895 to 1876) g | - | - | 0 | 0 | - | 7 | 5 | - | - | - | - | ||
| HPyV6/7Fi: TGGCACTTCAAYTGTGGTTG (from 738 to 757) h | - | - | 27 | 18 | - | 0 | 0 | - | - | - | - | ||
| HPyV6/7Ri: WCCAATKACATCCAAGGGGC (from 1730 to 1711) h | - | - | 16 | 5 | - | 0 | 0 | - | - | - | - | ||
| Expected amplicon size (bp) f | - | - | 881 | 893 | - | 993 | 1002 | - | - | - | - | ||
| HPyV | Genotype/Subtype/Subgroup (% total; % per Virus Species) | Accession Number |
|---|---|---|
| PyV1 | Ia (n = 2/2.74% Total; 13.33% HPyV1) | LC636348 a=5,c, LC636350 |
| Ib1 (n = 11/15.07% total; 73.33% HPyV1) | LC636351, LC636353, LC636359, LC636362, LC636363, LC636364, LC636374, LC636378, LC636381, LC636385, LC636400 | |
| III (n = 1/1.37% total; 6.67% HPyV1) | LC636340 | |
| I-like (n = 1/1.37% total; 6.67% HPyV1) | LC636403 | |
| HPyV2 | 1A (n = 3/4.11% total; 18.75% HPyV2) | LC636370, LC636395, LC636401 |
| 1B (n = 3/4.11% total; 18.75% HPyV2) | LC636349 a=5,c, LC636360 a=13,c, LC636404 | |
| 2A2 (n = 3/4.11% total; 18.75% HPyV2) | LC636358, LC636379, LC636380 | |
| 2B (n = 1/1.37% total; 6.25% HPyV2) | LC636396 | |
| 3A (n = 2/2.74% total; 12.50% HPyV2) | LC636377, LC636399 a=11,c | |
| 4 (n = 3/4.11% total; 18.75% HPyV2) | LC636357, LC636365 a=13,c, LC636394 | |
| 9 (n = 1/1.37% total; 6.25% HPyV2) | LC636376 | |
| HPyV5 | Africa (n = 8/10.96% total; 30.77% HPyV5) | LC636333, LC636335, LC636352, LC636355, LC636356, LC636384, LC636387 a=5,c, LC636398 |
| Europe/North America (n = 17/23.29% total; 65.38% HPyV5) | LC636341, LC636342, LC636343, LC636347, LC636354, LC636369, LC636373, LC636375, LC636386, LC636388 a=5,c, LC636389, LC636390, LC636391 a=5,c, LC636392, LC636393, LC636397, LC636402 | |
| Eu/NAm/Af (n = 1/1.37% total; 3.85% HPyV5) | LC636361 | |
| HPyV6 | n.a. b (n = 16/21.91% total; 100.00% HPyV6) | LC636334, LC636336, LC636337, LC636338, LC636339, LC636344, LC636345, LC636346, LC636366, LC636367, LC636368, LC636371, LC636372, LC636382, LC636383, LC636405 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Condez, A.C.; Nunes, M.; Filipa-Silva, A.; Leonardo, I.; Parreira, R. Human Polyomaviruses (HPyV) in Wastewater and Environmental Samples from the Lisbon Metropolitan Area: Detection and Genetic Characterization of Viral Structural Protein-Coding Sequences. Pathogens 2021, 10, 1309. https://doi.org/10.3390/pathogens10101309
Condez AC, Nunes M, Filipa-Silva A, Leonardo I, Parreira R. Human Polyomaviruses (HPyV) in Wastewater and Environmental Samples from the Lisbon Metropolitan Area: Detection and Genetic Characterization of Viral Structural Protein-Coding Sequences. Pathogens. 2021; 10(10):1309. https://doi.org/10.3390/pathogens10101309
Chicago/Turabian StyleCondez, Ana Carolina, Mónica Nunes, Andreia Filipa-Silva, Inês Leonardo, and Ricardo Parreira. 2021. "Human Polyomaviruses (HPyV) in Wastewater and Environmental Samples from the Lisbon Metropolitan Area: Detection and Genetic Characterization of Viral Structural Protein-Coding Sequences" Pathogens 10, no. 10: 1309. https://doi.org/10.3390/pathogens10101309