
Molecular Dynamics Study of Phosphorus Migration in Σ3(111) and Σ5(0-13) Grain Boundaries of α-Iron


Abstract
:
1. Introduction
2. Methods
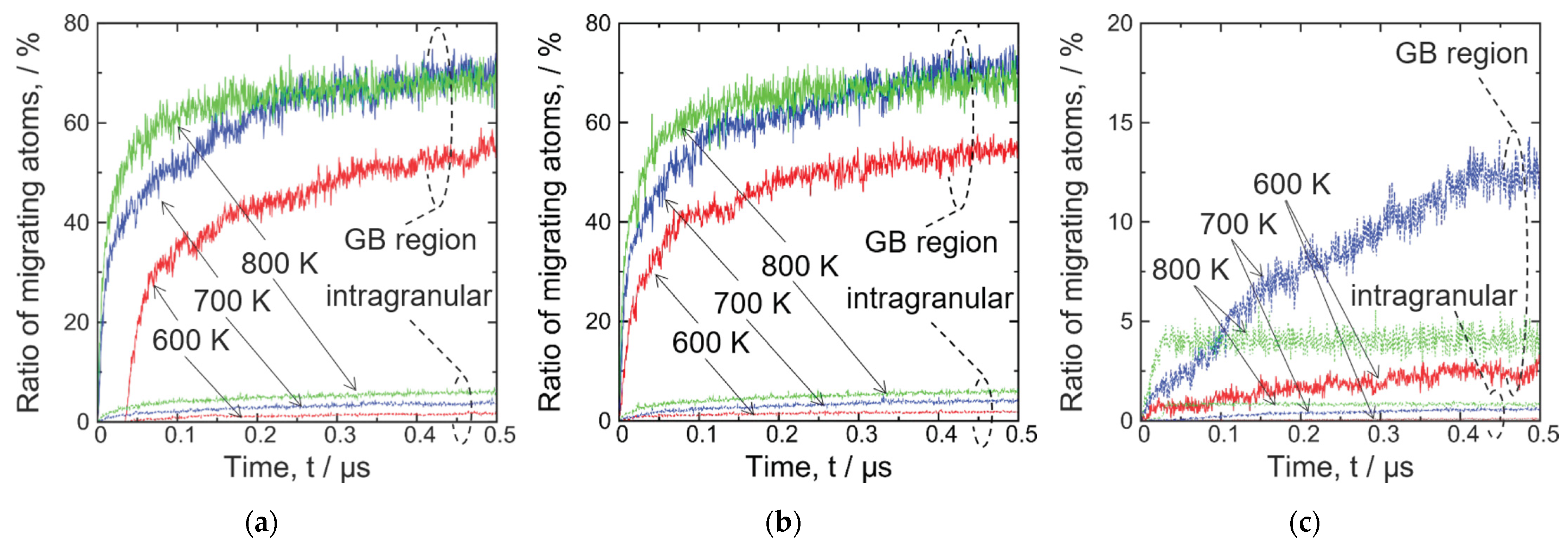
3. Results and Discussion
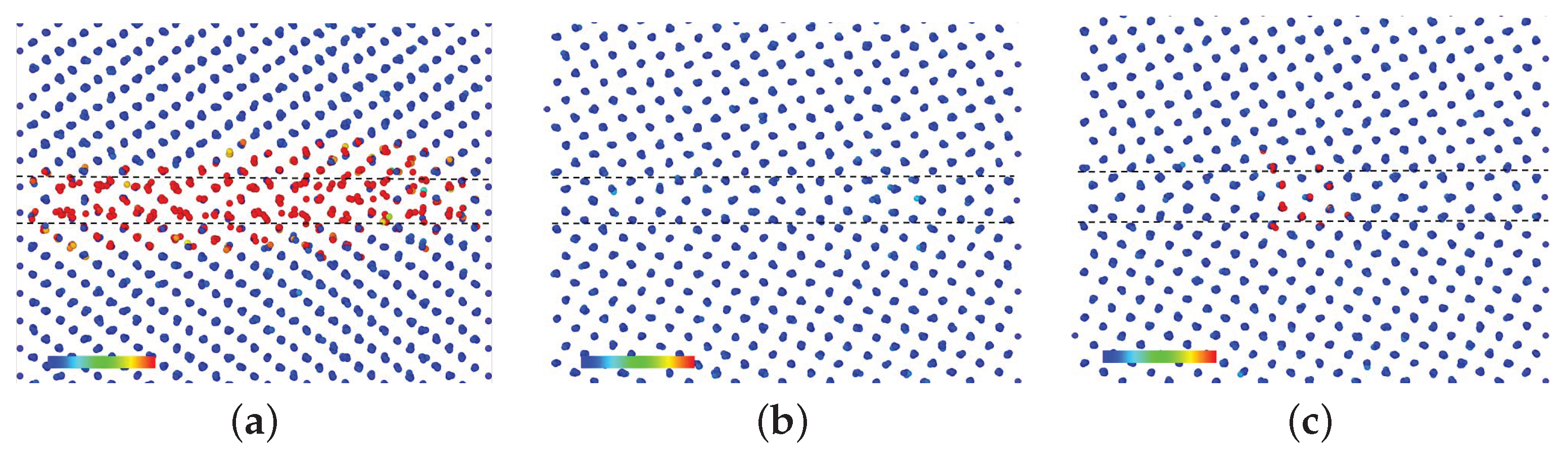
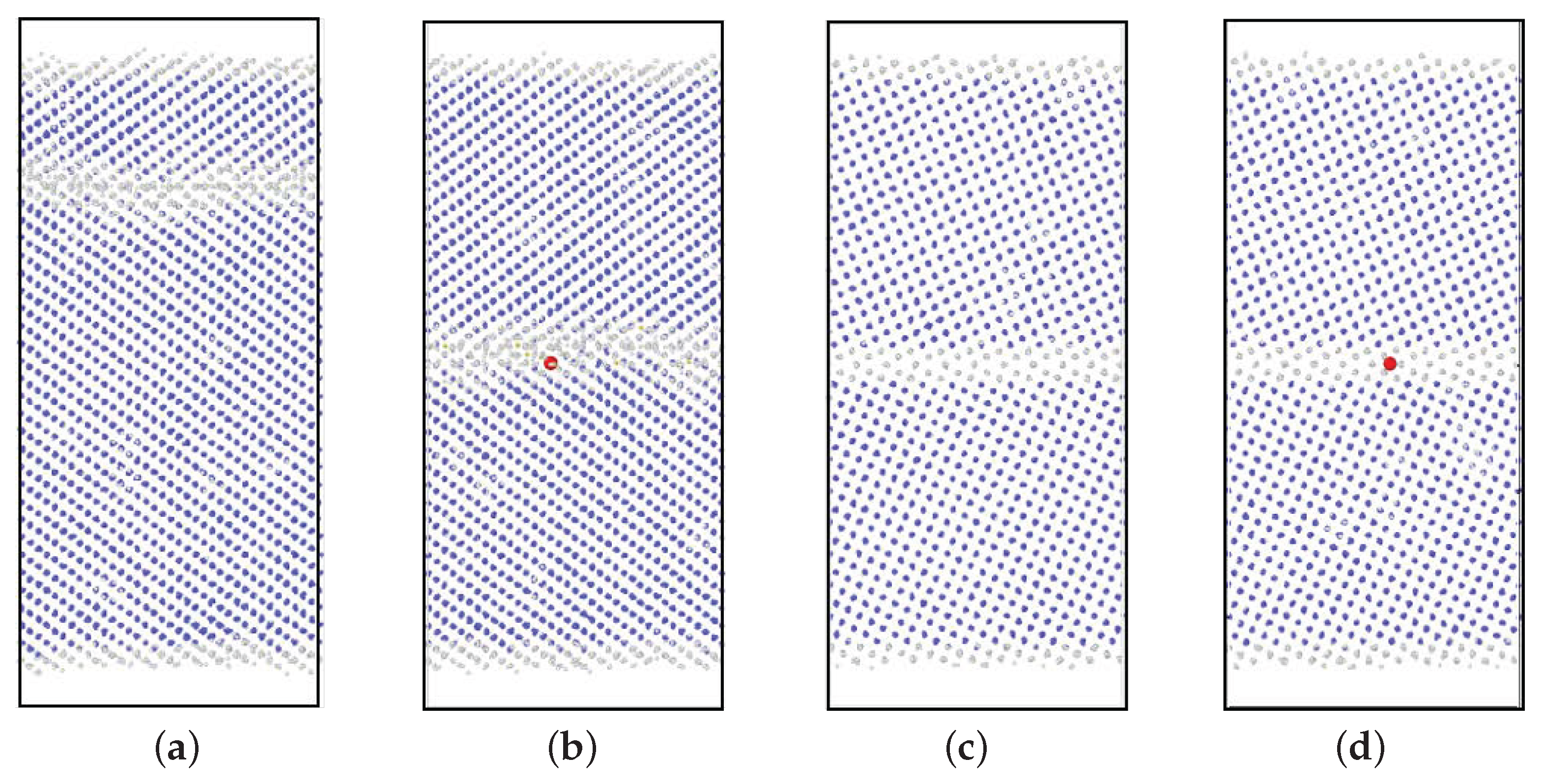
3.1. GBs without P Atoms
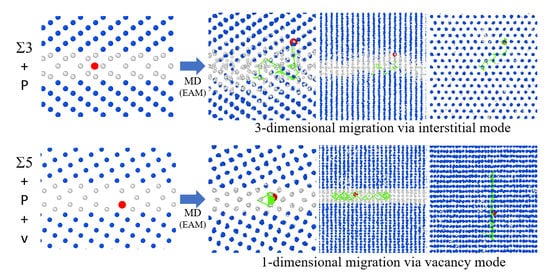
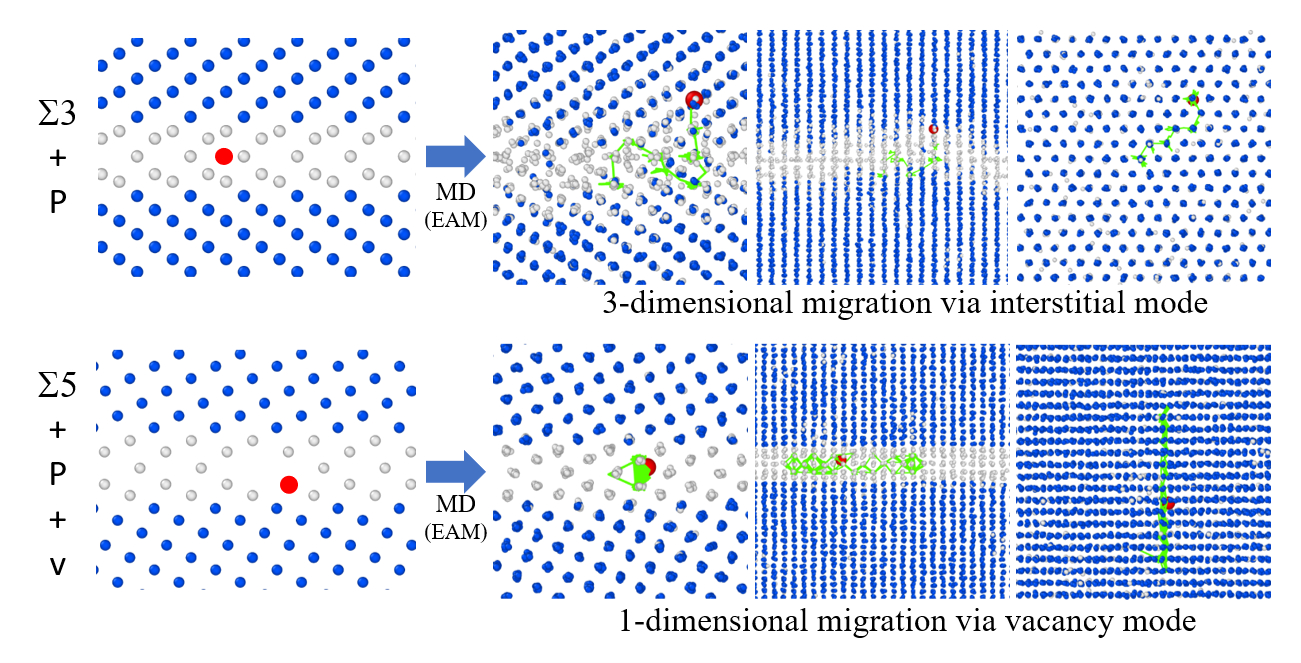
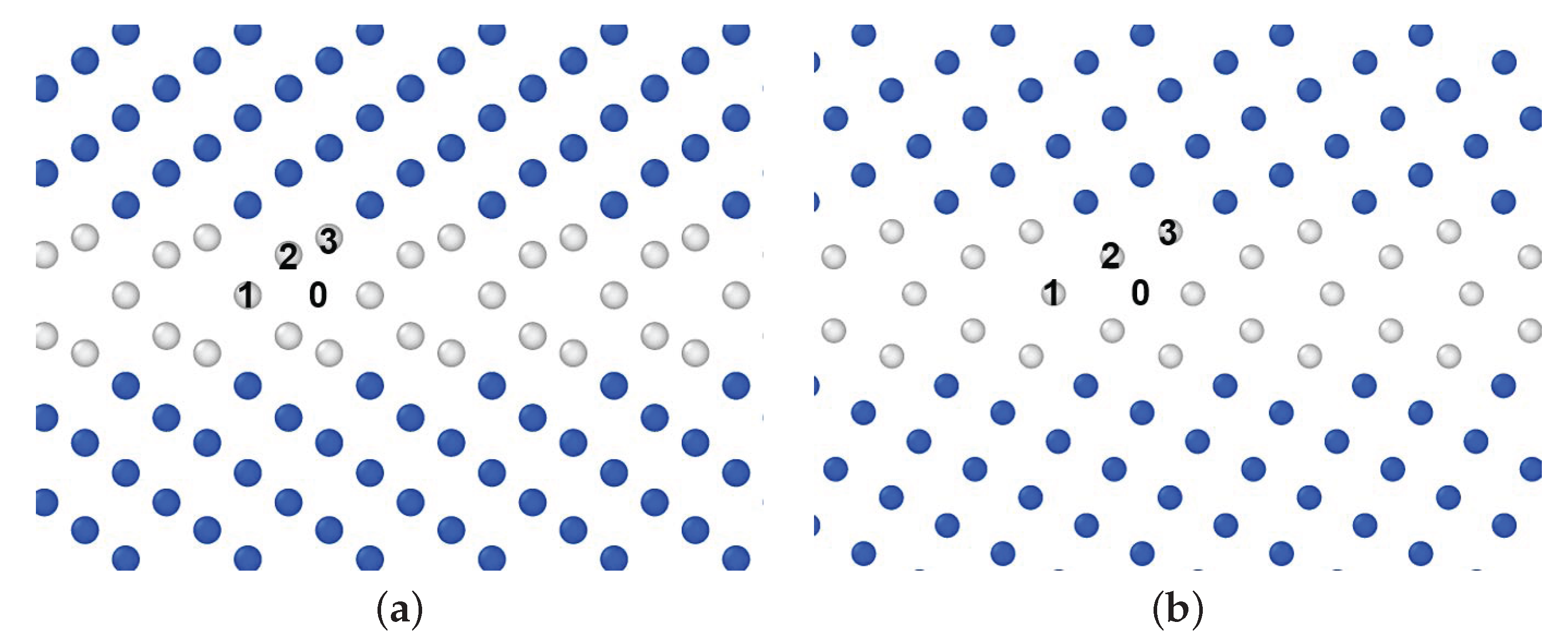
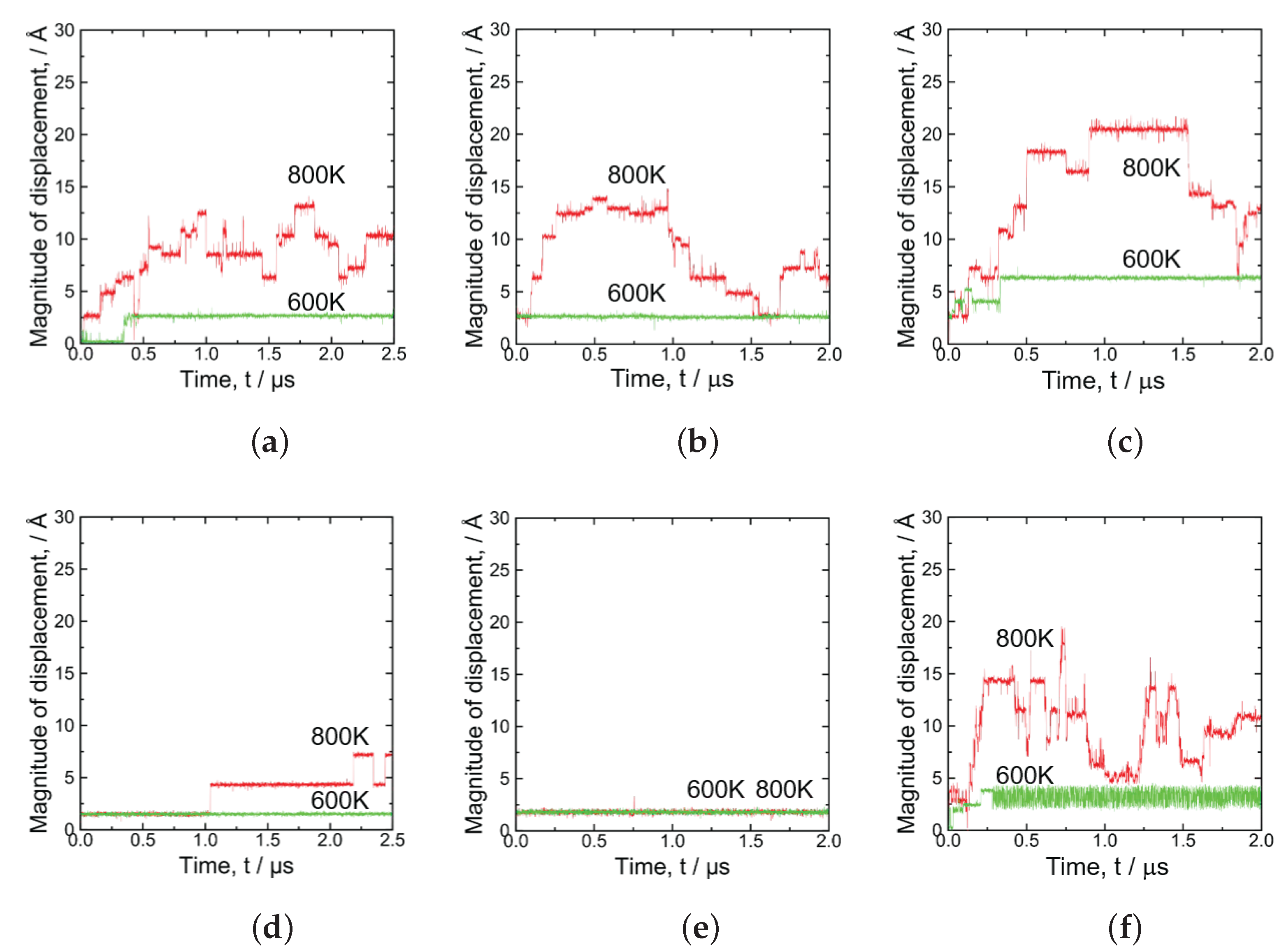
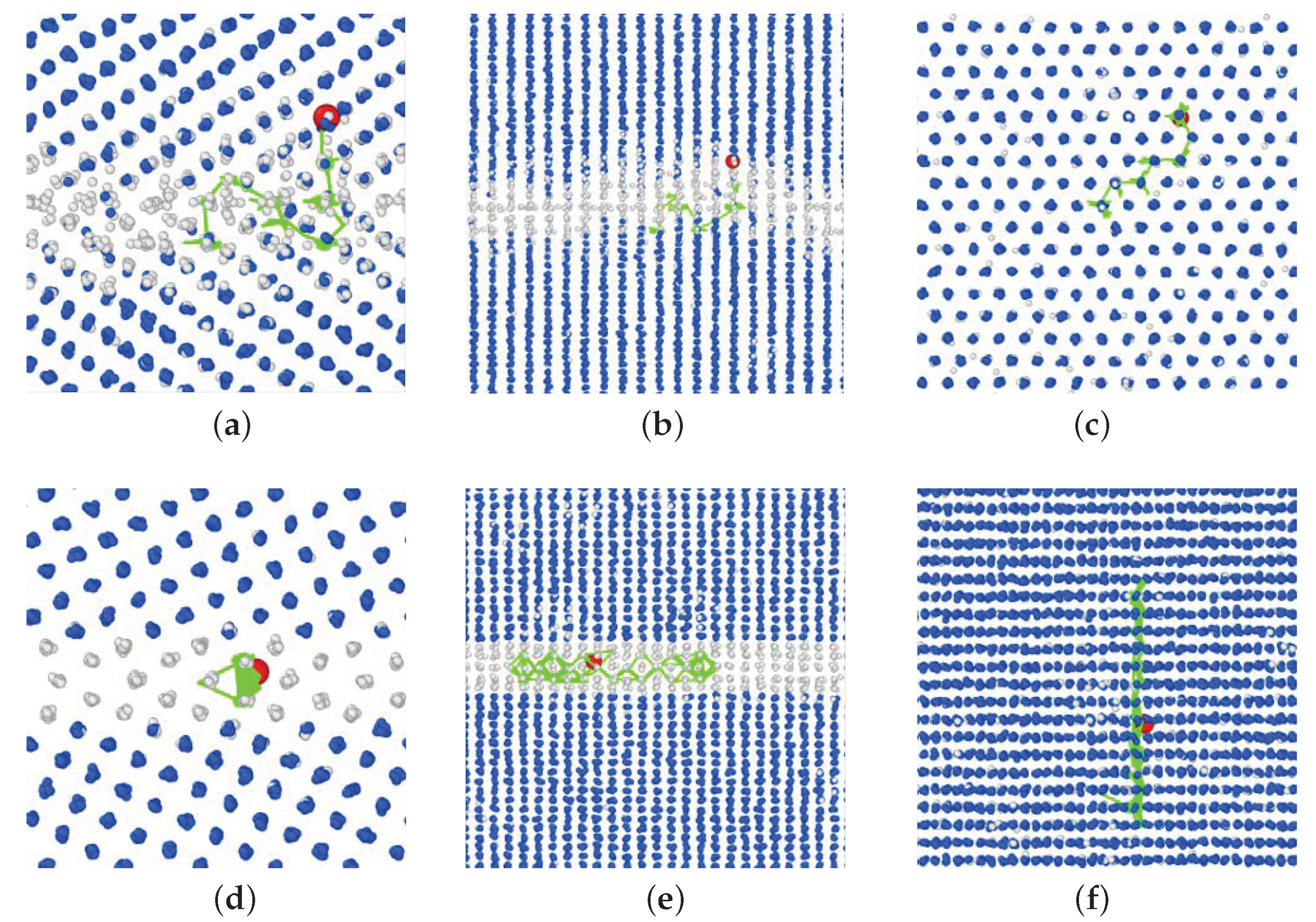
3.2. GBs with a P Atom
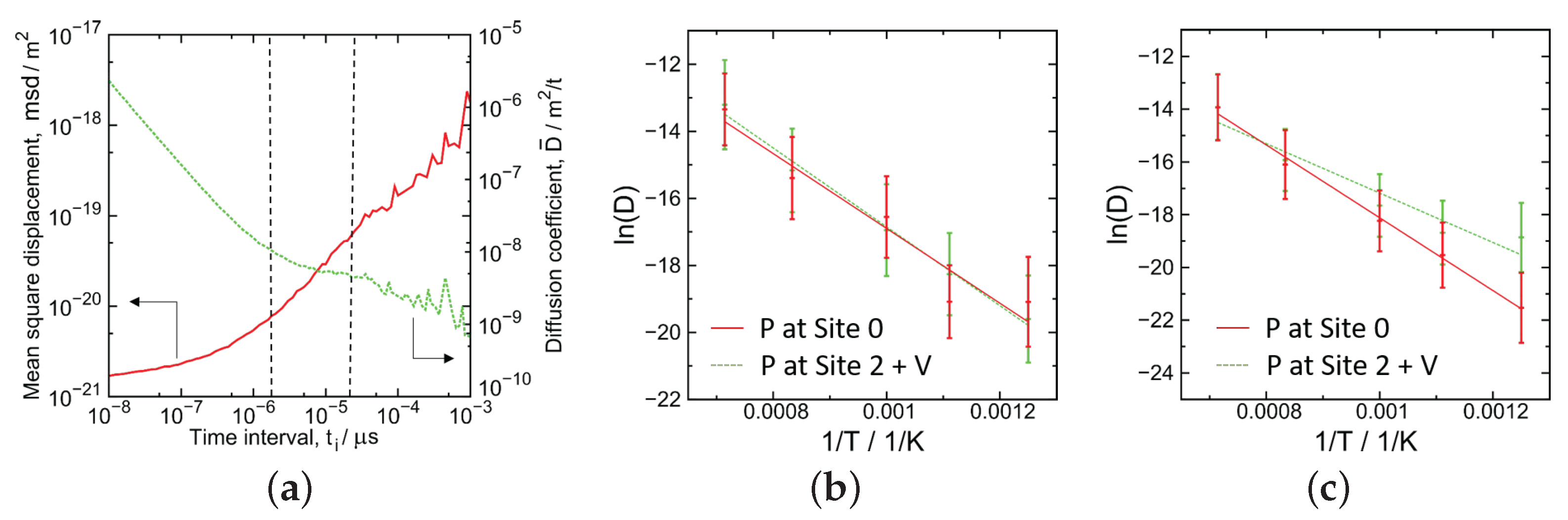
3.3. The Evaluation of Migration Barrier Energy
4. Concluding Remarks
- In the 3 GB, Fe atoms migrated at 600 K, 700 K, and 800 K, and the addition of a vacancy has no effect on the migration behavior at these temperatures. Moreover, in the 5 GB, no Fe atom migration was observed even at 800 K; migration occurs when a vacancy was added, but was less frequent than that in the case of 3 GB.
- The de-trapping of a vacancy from the GB region of the 5 GB was observed at 800 K.
- The P atom migrated frequently regardless of the initial site, that is substitutional or interstitial, in the 3 GB at 800 K. Moreover, in the 5 GB, the P migration was observed when a vacancy was placed at one of the nearest neighbor sites of the substitutional P atom at 800 K. In addition, the P atom migration was a one-dimensional motion in the 5 GB and a three-dimensional motion in the 3 GB.
- In the 3 GB, the de-trapping of the P atom from the GB region was observed at 800 K.
- The 3 GB undergoes significant thermal migration, which is suppressed by a P atom, while the 5 GB undergoes only slight thermal migration.
- The diffusion barrier energy of a P atom started from the interstitial site was compared with that of a P atom starting from a substitutional site with a vacancy nearby; the evaluated value of both cases was almost the same in the case of 3 GB, while in the case of 5 GB, the value of the former was larger than that of the latter.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kameda, J.; McMahon, C. The effects of Sb, Sn, and P on the strength of grain boundaries in a Ni-Cr Steel. Metall. Trans. A 1981, 12, 31–37. [Google Scholar] [CrossRef]
- Lejček, P.; Šob, M.; Paidar, V. Interfacial segregation and grain boundary embrittlement: An overview and critical assessment of experimental data and calculated results. Prog. Mater. Sci. 2017, 87, 83–139. [Google Scholar] [CrossRef]
- Druce, S.; Gage, G.; Jordan, G. Effect of ageing on properties of pressure vessel steels. Acta Metall. 1986, 34, 641–652. [Google Scholar] [CrossRef]
- Kameda, J.; Bloomer, T. Kinetics of grain-boundary segregation and desegregation of sulfur and phosphorus during post-irradiation annealing. Acta Mater. 1999, 47, 893–903. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Onizawa, K.; Suzuki, M.; Anderegg, J.; Nagai, Y.; Toyama, T.; Hasegawa, M.; Kameda, J. Effects of neutron-irradiation-induced intergranular phosphorus segregation and hardening on embrittlement in reactor pressure vessel steels. Acta Mater. 2008, 56, 4510–4521. [Google Scholar] [CrossRef]
- McMahon, C. Temper brittleness—An interpretive review. In Temper Embrittlement in Steel; ASTM International: West Conshohocken, PA, USA, 1968; pp. 127–167. [Google Scholar]
- Horn, R.; Ritchie, R.O. Mechanisms of tempered martensite embrittlement in low alloy steels. Metall. Trans. A 1978, 9, 1039–1053. [Google Scholar] [CrossRef]
- English, C.A.; Ortner, S.R.; Gage, G.; Server, W.L.; Rosinski, S.T. Review of phosphorus segregation and intergranular embrittlement in reactor pressure vessel steels. In Effects of Radiation on Materials: 20th International Symposium; ASTM International: West Conshohocken, PA, USA, 2001; pp. 151–173. [Google Scholar]
- Odette, G.; Lucas, G. Embrittlement of nuclear reactor pressure vessels. Jom 2001, 53, 18–22. [Google Scholar] [CrossRef]
- Druce, S.G.; English, C.A.; Foreman, A.J.; McElroy, R.J.; Vatter, I.A.; Bolton, C.J.; Buswell, J.T.; Jones, R.B. The modelling of irradiation-enhanced phosphorus segregation in neutron irradiated reactor pressure vessel submerged-arc welds. In Effects of Radiation on Materials: 17th International Symposium; ASTM International: West Conshohocken, PA, USA, 1996; pp. 119–137. [Google Scholar]
- Faulkner, R.G.; Little, E.A. Predictions of Interfacial Phosphorus Segregation in Irradiated Ferritic Steels. In Effects of Radiation on Materials: Sixteenth International Symposium; ASTM International: West Conshohocken, PA, USA, 1994; pp. 54–65. [Google Scholar]
- Murphy, S.; Perks, J. Analysis of phosphorus segregation in ion-irradiated nickel. J. Nucl. Mater. 1990, 171, 360–372. [Google Scholar] [CrossRef]
- Barashev, A. Segregation of phosphorus atoms to grain boundaries in ferritic steels under neutron irradiation. Philos. Mag. Lett. 2002, 82, 323–332. [Google Scholar] [CrossRef]
- Ebihara, K.; Suzudo, T.; Yamaguchi, M.; Nishiyama, Y. Introduction of vacancy drag effect to first-principles-based rate theory model for irradiation-induced grain-boundary phosphorus segregation. J. Nucl. Mater. 2013, 440, 627–632. [Google Scholar] [CrossRef]
- Ebihara, K.; Suzudo, T.; Yamaguchi, M. Modeling of Phosphorus Transport by Interstitial Dumbbell in α-Iron Using First-Principles-Based Kinetic Monte Carlo. Mater. Trans. 2017, 58, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Hata, K.; Takamizawa, H.; Hojo, T.; Ebihara, K.; Nishiyama, Y.; Nagai, Y. Grain-boundary phosphorus segregation in highly neutron-irradiated reactor pressure vessel steels and its effect on irradiation embrittlement. J. Nucl. Mater. 2021, 543, 152564. [Google Scholar] [CrossRef]
- Ebihara, K.; Suzudo, T. Atomistic simulation of phosphorus segregation to Σ3 (111) symmetrical tilt grain boundary in α-iron. Model. Simul. Mater. Sci. Eng. 2018, 26, 065005. [Google Scholar] [CrossRef]
- Azeem, M.M.; Wang, Q.; Zhang, Y.; Liu, S.; Zubair, M. Effect of Grain Boundary on Diffusion of P in Alpha-Fe: A Molecular Dynamics Study. Front. Phys. 2019, 7, 97. [Google Scholar] [CrossRef] [Green Version]
- Ebihara, K.; Suzudo, T. Molecular Dynamics Simulations of Phosphorus Migration in a Grain Boundary of α-Iron. In TMS 2020 149th Annual Meeting & Exhibition Supplemental Proceedings; Springer: Cham, Switzerland, 2020; pp. 995–1002. [Google Scholar]
- Honeycutt, J.D.; Andersen, H.C. Molecular dynamics study of melting and freezing of small Lennard-Jones clusters. J. Phys. Chem. 1987, 91, 4950–4963. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comp. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Ackland, G.J.; Mendelev, M.I.; Srolovitz, D.J.; Han, S.; Barashev, A. Development of an interatomic potential for phosphorus impurities in α-iron. J. Phys. Condens. Mater. 2004, 16, S2629–S2642. [Google Scholar] [CrossRef]
- Malerba, L.; Marinica, M.C.; Anento, N.; Björkas, C.; Nguyen, H.; Domain, C.; Djurabekova, F.; Olsson, P.; Nordlund, K.; Serra, A.; et al. Comparison of empirical interatomic potentials for iron applied to radiation damage studies. J. Nucl. Mater. 2010, 406, 19–38. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO—The Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012-1-7. [Google Scholar] [CrossRef]
- Suzudo, T.; Yamaguchi, M. Simulation of He embrittlement at grain boundaries in bcc transition metals. J. Nucl. Mater. 2015, 465, 695–701. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Nishiyama, Y.; Kaburaki, H. Decohesion of iron grain boundaries by sulfur or phosphorous segregation: First-principles calculations. Phys. Rev. B 2007, 76, 035418. [Google Scholar] [CrossRef]
- Gao, F.; Heinisch, H.; Kurtz, R.J. Diffusion of He interstitials in grain boundaries in α-Fe. J. Nucl. Mater. 2006, 351, 133–140. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GB | Energy [m] | Volume [m/m] |
|---|---|---|
| 1.30 | 3.08 | |
| 1.01 | 2.87 |
| [eV] | |||||
|---|---|---|---|---|---|
| GB | Method | Site 0 | Site 1 | Site 2 | Site 3 |
| DFT | 0.84 | 0.04 | 1.09 | 0.40 | |
| EAM | 0.55 | 0.22 | 1.17 | 0.44 | |
| DFT | 1.08 | −0.075 | 0.79 | 0.25 | |
| EAM | 0.89 | −0.077 | 0.69 | 0.28 |
| [eV] | ||
|---|---|---|
| GB | P at Site 0 | P at Site 2 with a Vacancy |
| 3 | 0.96 (±0.15) | 0.94 (±0.05) |
| 5 | 1.49 (±0.08) | 0.99 (±0.26) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebihara, K.-i.; Suzudo, T. Molecular Dynamics Study of Phosphorus Migration in Σ3(111) and Σ5(0-13) Grain Boundaries of α-Iron. Metals 2022, 12, 662. https://doi.org/10.3390/met12040662
Ebihara K-i, Suzudo T. Molecular Dynamics Study of Phosphorus Migration in Σ3(111) and Σ5(0-13) Grain Boundaries of α-Iron. Metals. 2022; 12(4):662. https://doi.org/10.3390/met12040662
Chicago/Turabian StyleEbihara, Ken-ichi, and Tomoaki Suzudo. 2022. "Molecular Dynamics Study of Phosphorus Migration in Σ3(111) and Σ5(0-13) Grain Boundaries of α-Iron" Metals 12, no. 4: 662. https://doi.org/10.3390/met12040662