
Crystal Chemistry and Electronic Properties of the Al-Rich Compounds, Al2Cu, ω-Al7Cu2Fe and θ-Al13Fe4 with Cu Solution



Abstract
:1. Introduction
2. Methods
3. Results
3.1. Effect of on-Site Coulomb Repulsion Correction on Cohesive Properties of Cu

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | LDA | LDA + U | GGA | GGA + U | Exper. (0 K) |
|---|---|---|---|---|---|
| Al: Face-Centered Cubic (FCC) lattice. Electronic configuration, [Ne] 3s2 3p¹ | |||||
| a (Å) | 3.984 (−1.2%) | - | 4.039 (+0.2%) | - | 4.0325 [45] |
| Ecoh (eV/Al) | −4.020 (+15.7%) | - | −3.511 (+3.6%) | - | −3.39 [46,53] |
| α-Fe: Body-Centered Cubic (BCC) lattice. Electronic configuration, [Ar] 3d6 4s2 | |||||
| a (Å) | 2.747 (−4.0%) | - | 2.831 (−1.0%) | - | 2.8607 [45] |
| Ecoh (eV/Fe) | −6.480 (+51.4%) | - | −4.933 (+15.3%) | - | −4.28 [46] |
| Cu: Face-Centered Cubic (FCC) lattice. Electronic configuration, [Ar] 3d¹⁰ 4s¹ | |||||
| a (Å) | 3.523 (−2.2%) 3.553 (−1.4%) [46] | 3.496 (−2.9%) | 3.635 (+0.9%) 3.630 (+0.7%) [30] 3.676 (+2.0%) [46] | 3.622 (+0.6%) | 3.6032 [45] |
| Ecoh (eV/Cu) | −4.676 (+34.4%) −4.353 [46] | −4.288 (+23.2%) | −3.501 (+0.6) −3.48 [30] | −3.079 (−11.5%) | −3.48 [30] −3.524 [54] |
| E (3d) (eV) | −1.78 | −2.49 | −1.65 | −2.34 | −2.4 (UPS) [51] −3.2 (XPS) [51] |
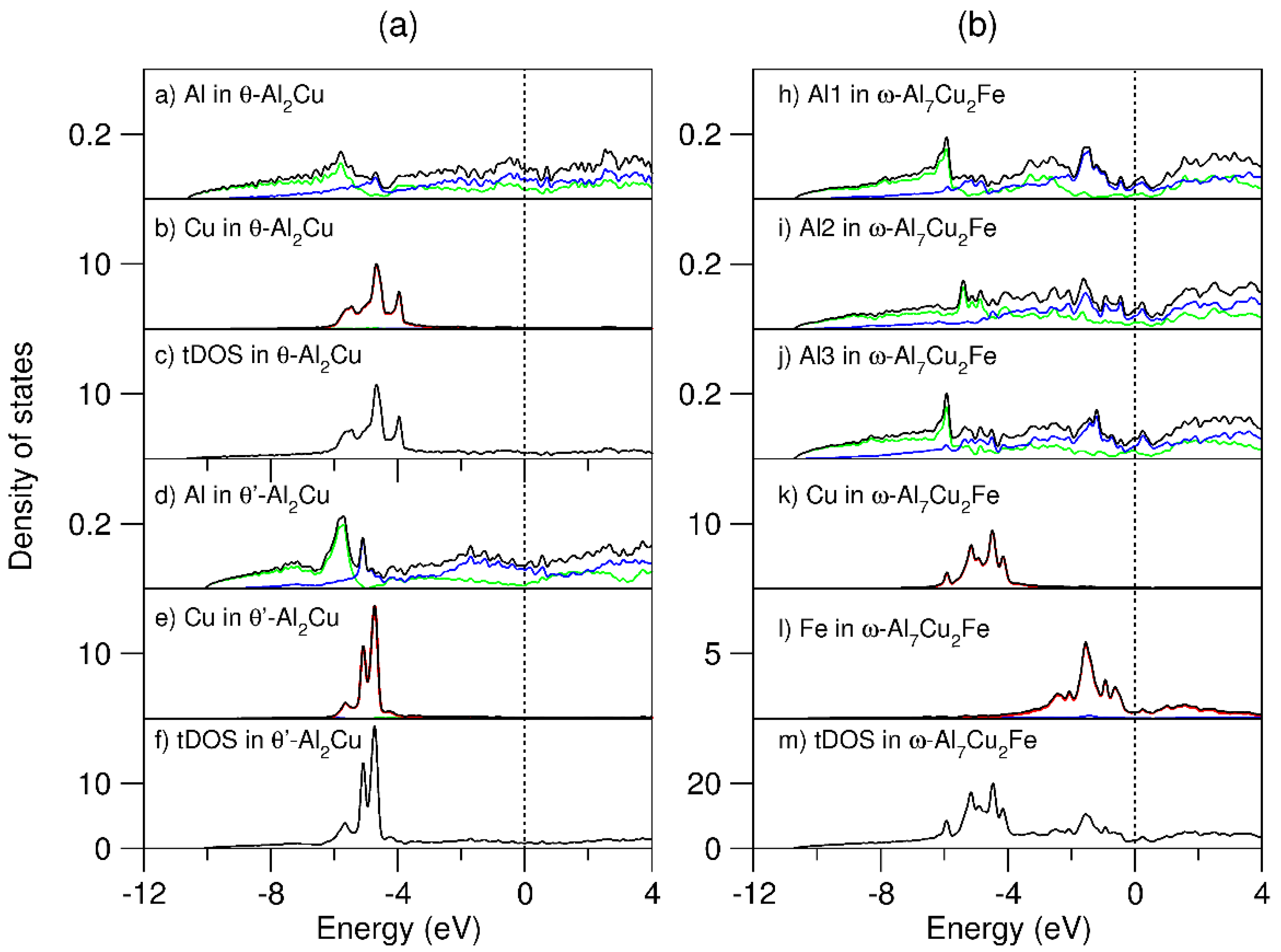
3.2. The Al-Rich Compounds θ-Al13Fe4, Al2Cu, and ω-Al7Cu2Fe in the Al-Fe-Cu System
| Compounds | Calculated Latt. Para. (Å)/Form. Energy | Experiments | |
|---|---|---|---|
| GGA | GGA + U | ||
| θ-Al13Fe4, Mon. | a = 15.426 | a = 15.492 | |
| C2/m, (nr. 12) | b = 8.022 | b = 8.078; | |
| c = 12.425 | c = 12.471 | ||
| β = 107.68 (°) | β = 107.69(°); | ||
| V = 1464.92 (Å3/cell) | V = 1486.88(Å3/cell) [22] | ||
| ΔEform = −0.329 eV/atom | ΔEform = −0.225 to −0.310 eV/atom [38,39,40] | ||
| θ-Al2Cu, Tetr. | a = 6.066 | 6.064 | a = 6.054 [57]; 6.063 [58];6.067 [59,60] |
| I4/mcm (Nr.140) | c = 4.874 | 4.864 | c = 4.864 [57]; 4.872 [58]; 4.877 [59,60] |
| V = 179.36 | 178.88(Å3/cell) | V = 178.27 (Å3/cell) [57],179.09 [58],179.52 [59,60] | |
| ΔEform = −0.157 | −0.125(eV/atom) | ||
| θ′-Al2Cu, Tetr. | a = 4.093 | 4.092 | a = 4.04 [60] |
| I-4m2 (Nr.119) | c = 5.786 | 5.782 | c = 5.80 [60] |
| V = 96.94 | 96.81(Å3/cell) | V = 94.67 (Å3/cell) [60] | |
| ΔEform = −0.176 | −0.124 (eV/atom) | ||
| ω-Al7Cu2Fe, Tetr. | a = 6.333 | 328 | a = 6.336 [61] 6.338 [62] |
| I-4m2 (Nr.119) | c = 14.763 | 14.773 | c = 14.870 [61] 14.832 [62] |
| V = 592.14 | 591.49 (Å3/cell) | V = 597.0 (Å3/cell) [61] 595.73 [62] | |
| ΔEform = −0.253 | −0.236 (eV/atom) | ||

3.3. Cu Substitution at the Atomic Sites in θ-Al13Fe4
| Spe., Site (sym) | Al/Fe-M in Pure θ-Al13Fe4 | Cu-M in Doped θ-Al13Fe4 | ΔEsub (eV/Cell) |
|---|---|---|---|
| Al1, 4i (m) | Al1-7Al(2.54–2.89) -3Fe: 2.45, 2.53, 2.62 | Cu-7Al(2.44–2.84) -3Fe: 2.49, 2.66, 2.79 | 0.637 |
| Al2, 4i (m) | Al2-4Al(2.92–2.97) -2Fe: 2.36(×2) | Cu-4Al(2.76–2.88) -2Fe: 2.49(×2) | 0.801 |
| Al3, 4i (m) | Al3-10Al(2.71–2.96) -2Fe: 2.44, 2.52 | Cu-10Al(2.64–2.85) -2Fe: 2.53, 2.62 | 0.448 |
| Al4, 4i (m) | Al4-7Al(2.53–2.75) -4Fe: 2.51, 2.61(×2), 2.63 | Cu-7Al(2.41–2.68) -4Fe:2.62, 2.65(×2), 2.74 | 0.453 |
| Al5, 4i (m) | Al5-8Al(2.67–2.86) -2Fe: 2.41, 2.44 | Cu-10Al(2.62–2.84) -2Fe: 2.50, 2.52 | 0.422 |
| Al6, 4i (m) | Al6-7Al(2.56–2.86) -3Fe: 2.47, 2.48, 2.55 | Cu-7Al(2.48–2.79) -3Fe: 2.52, 2.58, 2.70 | 0.805 |
| Al7, 2d (2/m) | Al7-8Al(2.69–2.80) -2Fe: 2.47(×2) | Cu-8Al(2.58–2.69) -2Fe: 2.59(×2) | −0.123 |
| Al8, 4i (m) | Al8-7Al(2.56–2.81) -4Fe: 2.47(×2), 2.61, 2.70 | Cu-7Al(2.41–2.68) -4Fe:2.55(×2), 2.69, 2.77 | 0.646 |
| Al9, 4i (m) | Al9-7Al(2.53–2.67) -4Fe: 2.46, 2.49(×2), 2.86 | Cu-7Al(2.39–2.57) -4Fe: 2.57, 2.58(×2), 2.89 | 0.246 |
| Al10, 8j (1) | Al10-9Al(2.67–2.86) -2Fe: 2.48, 2.52 | Cu-9Al(2.59–2.84) -3Fe: 2.58, 2.62, 2.84 | 0.453 |
| Al11, 8j (1) | Al11-9Al(2.63–2.97) -3Fe: 2.46, 2.51, 2.77 | Cu-9Al(2.54–2.82) -3Fe: 2.56, 2.62, 2.87 | 0.387 |
| Al12, 8j (1) | Al12-8Al(2.64–2.96) -3Fe: 2.46, 2.56, 2.64 | Cu-9Al(2.59–2.96) -3Fe: 2.55, 2.62, 2.85 | 0.586 |
| Al13, 8j (1) | Al13-7Al(2.67–2.88) -3Fe: 2.44, 2.55, 2.58 | Cu-8Al(2.60–2.97) -3Fe: 2.52, 2.59, 2.75 | 0.661 |
| Al14, 8j (1) | Al14-8Al(2.64–2.92) -3Fe: 2.45, 2.46, 2.81 | Cu-9Al(2.38–2.70) | 2.074 |
| Al15, 8j (2) | Al15-8Al(2.78–2.89) -4Fe: 2.46(×4) | Cu-8Al(2.72–2.81) -4Fe: 2.51(×4) | 0.879 |
| Fe1, 4i (m) | Fe1-11Al(2.46–2.86) | Cu-11Al(2.51–2.93) | 0.548 |
| Fe2, 4i (m) | Fe2-10Al(2.44–2.70) | Cu-10Al(2.49–2.85) | 0.548 |
| Fe3, 4i (m) | Fe3-10Al(2.44–2.77) -1Fe: 2.91 | Cu-10Al(2.50–2.88) -1Fe: 2.86 | 0.638 |
| Fe4, 4i (m) | Fe4-10Al(2.41–2.72) -1Fe: 2.93 | Cu-10Al(2.47–2.77) -1Fe: 2.91 | 0.672 |
| Fe5, 8j (1) | Al1-9Al(2.36–2.61) | Cu-9Al(2.37–2.70) | 1.004 |

3.4. Crystal and Electronic Properties of θ-Al76Cu2Fe24


4. Discussion
5. Summary
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mondolfo, L.F. Aluminium Alloys: Structure and Properties; Butterworth & Co., Ltd.: London, UK, 1976. [Google Scholar]
- Zhang, L.F.; Gao, J.W.; Nana, L.; Damoah, W.; Robertson, D.G. Removal of iron aluminum: A review. Miner. Process. Extr. Metall. Rev. 2012, 33, 99–157. [Google Scholar] [CrossRef]
- Que, Z.P.; Wang, Y.; Fan, Z. Formation of the Fe-containing intermetallic compounds during solidification of Al-5Mg-2Si-0.7Mn-1.1Fe alloy. Metal. Mater. Trans. A 2018, 49, 2173–2181. [Google Scholar] [CrossRef] [Green Version]
- Que, Z.P.; Mendis, C.L. Formation of theta-Al13Fe4 and the multi-step phase transformation to lapha-Al8Fe2Si, beta-Al5FeSi and delta-Al4FeSi2 in Al-20Si-0l7Fe alloy. Intermetallics 2020, 127, 106960. [Google Scholar] [CrossRef]
- Cai, Q.; Mendis, C.L.; Wang, S.A.; Chang, I.T.H.; Fan, Z. Effect of heat treatment on microstructure and tensile properties of die-cast Al-Cu-Si-Mg alloys. J. Alloys Compd. 2021, 881, 160559. [Google Scholar] [CrossRef]
- Barbaux, Y.; Pons, G. New rapidly solidified aluminium alloys for elevated temperature applications on aerospace structures. J. Phys. IV 1993, 3, 191–196. [Google Scholar] [CrossRef]
- Guinier, A.; Preston, G.D. Structure of age-hardened aluminium-copper alloys. Nature 1938, 142, 569–570. [Google Scholar] [CrossRef]
- Schlesinger, M.E. Aluminum Recycling; Taylor & Francis Group: Abingdon-on-Thames, UK, 2017. [Google Scholar]
- Gesing, A.; Berry, L.; Dalton, R.; Wolanski, R. Assuring continued recyclability of automotive aluminum alloys: Grouping of wrought alloys by color, X-ray adsorption and chemical composition-based sorting. In Automotive Alloys and Aluminum Sheet and Plate Rolling and Finishing Technology Symposia, Proceedings of the TMS 2002 Annual Meeting and Exibition, Seattle, WA, USA, 18–21 February 2002; Minerals, Metals and Materials Society (TMS): Warrendale, PA, USA; pp. 3–17.
- Fang, X.; Shao, G.; Liu, Y.Q.; Fan, Z. Effects of intensive forced melt convection on the mechanical properties of Fe-containing Al-Si based alloys. Mater. Sci. Eng. A 2007, 445, 65–72. [Google Scholar] [CrossRef]
- Sundman, B.; Ohnuma, I.; Dupin, N.; Kattner, U.R.; Fries, S.G. An assessment of the entire Al-Fe system including D0(3) ordering. Acta Mater. 2009, 57, 2896–2908. [Google Scholar] [CrossRef]
- Zienert, T.; Fabrichnaya, O. Experimental investigation and thermodynamic assessment of the Al-Fe system. J. Alloys Compd. 2018, 743, 795–811. [Google Scholar] [CrossRef]
- Li, X.L.; Scherf, A.; Heilmaier, M.; Stein, F. The Al-rich part of the Fe-Al phase diagram. JPEDAV 2016, 37, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Shabestari, S.G. The effect of iron and manganese on the formation of intermetallic compounds in aluminum-silicon alloys. Mater. Sci. Eng. A 2004, 383, 289–298. [Google Scholar] [CrossRef]
- Malakhov, D.V.; Panahi, D.; Gallernault, M. On the formation of metallics in rapidly solidifying Al-Fe-Si alloys. CALPHAD 2010, 34, 159–166. [Google Scholar] [CrossRef]
- Taylor, J.A. Iron-containing intermetallic phases in Al-Si based casting alloys. Procedia Mater. Sci. 2012, 1, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Raghavan, V. Al-Cu-Fe. JPEDAV 2010, 31, 449–452. [Google Scholar] [CrossRef]
- Mehta, U.; Yadav, S.K.; Koirala, I.; Adhikari, D. Thermo-physical properties of ternary Al-Cu-Fe alloy in liquid state. Philos. Mag. 2020, 100, 2417–2435. [Google Scholar] [CrossRef]
- Zhu, L.L.; Sato-Medina, S.; Cuadrado-Castillo, W.; Henning, R.G.; Manual, M.V. New experimental studies on the phase diagram of the Al-Cu-Fe quasicrystal-forming system. Mater. Des. 2019, 185, 108186. [Google Scholar] [CrossRef]
- Corby, R.N.; Black, P.J. The structure of α–(AlFeSi) by anomalous-dispersion methods. Acta Cryst. B 1977, 33, 3468–3475. [Google Scholar] [CrossRef]
- Cooper, M. The crystal structure of ternary alpha-(Al Fe Si). Acta Cryst. 1967, 23, 1106–1107. [Google Scholar] [CrossRef]
- Grin, Y.; Burkhard, U.; Ellner, M.; Peters, K. Refinement of the Fe4Al13 structure and its relationship to the quasihomogical homeotypical structures. Z. Krist. 1994, 209, 479–487. [Google Scholar]
- Popčević, P.; Smontara, A.; Ivkov, J.; Wencka, M.; Komelj, M.; Jeglič, P.; Vrtnik, S.; Bohnar, M.; Jagličić, Z.; Baer, B.; et al. Anisotropic physical properties of the Al13Fe4 complex intermetallic and its ternary derivative Al13(Fe, Ni)4. Phys. Rev. B 2010, 81, 184203. [Google Scholar] [CrossRef]
- Jeglič, C.; Vrtnik, S.; Bobnar, M.; Klanjček, M.; Bauer, B.; Gille, P.; Grin, Y.; Haarmann, F.; Dolinček, J. M-Al-M groups trapped in cages of Al13M4 (M = Co, Fe, Ni, Ru) complex intermetallic phases as seen via NMR. Phys. Rev. B 2010, 82, 104201. [Google Scholar] [CrossRef]
- Freiburg, C.; Grushko, B. An Al13Fe4 phase in the Al-Cu-Fe alloy system. J. Alloys Compd. 1994, 210, 149–152. [Google Scholar] [CrossRef]
- Genba, M.; Sugiyama, K.; Hiraga, K.; Yokoyama, Y. Crystalline structure of a Cu-substituted λ-Al13Fe4 phase by means of the anomalous X-ray scattering. J. Alloys Compd. 2002, 342, 143–147. [Google Scholar] [CrossRef]
- Wang, J.; Shang, S.L.; Wang, Y.; Mei, Z.G.; Liang, Y.F.; Du, Y.; Liu, Z.K. First-principles calculations of binary compounds: Enthalpies of formation and elastic properties. CALPHAD 2011, 35, 562–573. [Google Scholar] [CrossRef]
- Fang, C.M.; Que, Z.P.; Fan, Z. Crystal Chemistry and Electronic Structure of the β-AlFeSi phase from First-Principles. J. Solid State Chem. 2021, 299, 122199. [Google Scholar] [CrossRef]
- Staab, T.E.M.; Folegati, P.; Wolfertz, I.; Puska, M.J. Stability of Cu-precipitates in Al-Cu alloys. Appl. Sci. 2018, 8, 1003. [Google Scholar] [CrossRef] [Green Version]
- Janthon, P.; Luo, S.J.; Kozlov, S.M.; Viñes, F.; Limtrakul, J.; Truhlar, D.G.; Illas, F. Bulk properties of transition metals: A challenge for the design of universal density functional. J. Chem. Theory Comput. 2014, 10, 3832–3839. [Google Scholar] [CrossRef] [Green Version]
- Zienert, T.; Leineeber, A.; Fabrichnaya, O. Heat capacity of Fe-Al intermetallics: B2-FeAl, FeAl2, Fe2Al5 and Fe4Al13. J. Alloys Compd. 2017, 725, 848–859. [Google Scholar] [CrossRef]
- van Alboom, A.; Lemmens, B.; Breitbach, B.; de Grave, E.; Cottenier, S.; Verbeken, K. Multi-method identification and characterization of the intermetallic surface layers of Al-coated steel: FeAl3 or Fe4Al13 and Fe2Al5 or Fe2Al5+x. Surf. Coat. Technol. 2017, 324, 419–428. [Google Scholar] [CrossRef]
- Ledieu, J.; Gaudry, E.; Loli, L.N.; Villaseca, S.A.; de Weerd, M.C.; Hahne, M.; Gille, P.; Dubois, J.M.; Fournée, V. Structural investigation of the (010) surface of the Al13Fe4 catalyst. Phys. Rev. Lett. 2013, 110, 076102. [Google Scholar] [CrossRef] [Green Version]
- Armbrüster, M.; Kovnir, K.; Friedrich, M.; Teschner, D.; Wowsnick, G.; Hahne, M.; Gille, P.; Szentmiklósi, L.; Feuerbacher, M.; Heggen, M.; et al. Al13Fe4 as a low-cost alternative for palladium in heterogeneous hydrogenation. Nat. Mater. 2012, 11, 690–693. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.M.; Dinsdale, A.; Que, Z.P.; Fan, Z. Intrinsic defects in and electronic properties of θ-Al13Fe4: An ab initio DFT study. J. Phys. Mater. 2019, 2, 015004. [Google Scholar] [CrossRef]
- Dinsdale, A.; Fang, C.M.; Que, Z.P.; Fan, Z. Understanding the thermodynamics and crystal structure of complex Fe containing intermetallic phases formed on solidification of aluminium alloys. JOM 2019, 71, 1731–1736. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.M.; Que, Z.P.; Dinsdale, A.; Fan, Z. Si solution in θ-Al13Fe4 from first-principles. Intermetallics 2020, 126, 106939. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17978. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.M.; Sluiter, M.H.F.; van Huis, M.A.; Ande, C.K.; Zandbergen, H.W. Origin of predominance of cementite among iron carbides in steel at elevated temperature. Phys. Rev. Lett. 2010, 105, 055503. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Wyckoff, R.W.G. The Structure of Crystals; Reinhold Publishing Corporation: New York, NY, USA, 1963. [Google Scholar]
- Arblaster, J. Selected Values of the Crystallographic Properties of the Elements; ASM International: Materials Park, OH, USA, 2018. [Google Scholar]
- Connelly, N.G.; Damhus, T.; Hartshorn, R.M.; Hutton, A.T. Nomenclature of Inorganic Chemistry: IUPAC Recommendations; RSC Publishing: Cambridge, UK, 2005. [Google Scholar]
- Hubbard, J. Electron correlations in narrow energy bands. Proc. R. Soc. Lond. Ser. A 1963, 276, 238–257. [Google Scholar]
- Lichtenstein, A.I.; Anisimov, V.I.; Zaanen, Z. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 1995, 52, R5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Maxisch, T.; Ceder, G. Oxidation energies of transition metal oxides within the GGA + U framework. Phys. Rev. B 2006, 73, 195107. [Google Scholar] [CrossRef] [Green Version]
- Souissi, M.; Fang, C.M.; Sahara, R.; Fan, Z. Formation energies of θ-Al2Cu phase and precursor Al-Cu compounds: Importance of on-site Coulomb repulsion. Comput. Mater. Sci. 2021, 194, 110461. [Google Scholar] [CrossRef]
- Fadley, C.S.; Shirley, D.A. Electronic densities of states from X-ray photoelectron spectroscopy. J. Res. Natl. Bureau Stand. A 1970, 74, 543–558. [Google Scholar] [CrossRef]
- Şaşioğlu, E.; Friedrich, C.; Blügel, S. Effective Coulomb interaction in transition metals from constrained random-phase approximation. Phys. Rev. B 2011, 83, 121101. [Google Scholar] [CrossRef] [Green Version]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Ambrosetti, A.; Silvestrelli, P.L. Cohesive properties of noble metals by van der Waals-corrected density functional theory: Au, Ag, and Cu as case studies. Phys. Rev. B 2016, 94, 045124. [Google Scholar] [CrossRef] [Green Version]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef] [Green Version]
- Paier, J.; Marsman, M.; Hummer, K.; Kresse, G.; Gerber, I.C.; Angyan, J.G. Screened hybrid density functionals applied to solids. J. Chem. Phys. 2006, 124, 154709. [Google Scholar] [CrossRef] [Green Version]
- Bradley, A.J.; Jones, P. An X-ray investigation of the copper-aliminium alloys. J. Inst. Met. 1933, 625, 131–162. [Google Scholar]
- Havinga, E.E.; Damsma, H.; Hokkeling, P. Compounds and pseudo-binary alloys with the CuAl2(Cl6)-type structure. I. Preparation and X-ray results. J. Less-Common Met. 1972, 27, 169–186. [Google Scholar] [CrossRef]
- Meetsma, A.; de Boer, J.L.; van Smaalen, S. Refinement of the crystal structure of tetragonal Al2C. J. Solid State Chem. 1989, 370, 370–372. [Google Scholar] [CrossRef]
- Wang, S.; Fan, C. Crystal structures of Al2Cu revisited: Understanding existing phases and exploring other potential phases. Metals 2019, 9, 1037. [Google Scholar] [CrossRef] [Green Version]
- Bown, M.; Bown, P. The structure of FeCu2Al7 and T(CoCuAl). Acta Cryst. 1956, 9, 911–914. [Google Scholar] [CrossRef]
- Tian, J.Z.; Zhao, Y.H.; Wen, Z.Q.; Hou, H.; Han, P.D. Physical properties and Debye temperature of Al7Cu2Fe alloy under various pressures analyzed by first-principles. Solid State Commun. 2017, 257, 6–10. [Google Scholar] [CrossRef]



| Compound | θ-Al78Fe24 | θ-Al76Cu2Fe24 | |||||
|---|---|---|---|---|---|---|---|
| Lattice Parameters (Å) | a = 15.426, b = 8.022, c = 12.425; | a = 15.504, b = 7.930, c = 12.459; | |||||
| Β = 107.68(°), V = 1464.92(Å3) | Β = 108.16 (°), V = 1455.62(Å3) | ||||||
| ΔEform (eV/atom) | −0.329 | −0.332 | |||||
| Species | site | x | y | z | x | y | z |
| Al1 | 4i | 0.065 | 0 | 0.173 | 0.066 | 0 | 0.1727 |
| Al2 | 4i | 0.323 | 0 | 0.2812 | 0.3214 | 0 | 0.2786 |
| Al3 | 4i | 0.2371 | 0 | 0.5354 | 0.2334 | 0 | 0.5362 |
| Al4 | 4i | 0.0737 | 0 | 0.5803 | 0.0738 | 0 | 0.5794 |
| Al5 | 4i | 0.2409 | 0 | 0.9613 | 0.2391 | 0 | 0.9557 |
| Al6 | 4i | 0.4788 | 0 | 0.83 | 0.4778 | 0 | 0.8312 |
| Al7 | 2d | 0.5 | 0 | 0.5 | 0.5 | 0 | 0.5000 * |
| Al8 | 4i | 0.3049 | 0 | 0.7726 | 0.305 | 0 | 0.7715 |
| Al9 | 4i | 0.0874 | 0 | 0.7883 | 0.0826 | 0 | 0.7845 |
| Al10 | 8j | 0.1854 | 0.2175 | 0.1107 | 0.1859 | 0.2166 | 0.1108 |
| Al11 | 8j | 0.368 | 0.2117 | 0.1097 | 0.3682 | 0.2111 | 0.1102 |
| Al12 | 8j | 0.1782 | 0.2193 | 0.3345 | 0.1787 | 0.2187 | 0.3366 |
| Al13 | 8j | 0.4922 | 0.2333 | 0.33 | 0.4901 | 0.2222 | 0.3352 |
| Al14 | 8j | 0.3631 | 0.2186 | 0.4781 | 0.367 | 0.2064 | 0.4768 |
| Al15 | 4g | 0 | 0.2503 | 0 | 0 | 0.2501 | 0 |
| Fe1 | 4i | 0.0853 | 0 | 0.383 | 0.0836 | 0 | 0.3812 |
| Fe2 | 4i | 0.4015 | 0 | 0.6237 | 0.396 | 0 | 0.6298 |
| Fe3 | 4i | 0.0908 | 0 | 0.9878 | 0.0904 | 0 | 0.9872 |
| Fe4 | 4i | 0.402 | 0 | 0.9851 | 0.4022 | 0 | 0.9861 |
| Fe5 | 8j | 0.3198 | 0.2935 | 0.2779 | 0.3205 | 0.2942 | 0.2774 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, C.; Souissi, M.; Que, Z.; Fan, Z. Crystal Chemistry and Electronic Properties of the Al-Rich Compounds, Al2Cu, ω-Al7Cu2Fe and θ-Al13Fe4 with Cu Solution. Metals 2022, 12, 329. https://doi.org/10.3390/met12020329
Fang C, Souissi M, Que Z, Fan Z. Crystal Chemistry and Electronic Properties of the Al-Rich Compounds, Al2Cu, ω-Al7Cu2Fe and θ-Al13Fe4 with Cu Solution. Metals. 2022; 12(2):329. https://doi.org/10.3390/met12020329
Chicago/Turabian StyleFang, Changming, Maaouia Souissi, Zhongping Que, and Zhongyun Fan. 2022. "Crystal Chemistry and Electronic Properties of the Al-Rich Compounds, Al2Cu, ω-Al7Cu2Fe and θ-Al13Fe4 with Cu Solution" Metals 12, no. 2: 329. https://doi.org/10.3390/met12020329