
Transient Liquid Phase Sintering of PM Steel—A Matter of the Heating Rate


Abstract
:1. Introduction
2. Materials and Methods
2.1. Evolution of the Melting Process at Fast Heating and Cooling Rates
2.2. Influence of the Heating Rate on the Melting Process
2.3. Hardenability of Samples Sintered at 1180 °C with 10 K/min Heating Rate
3. Results
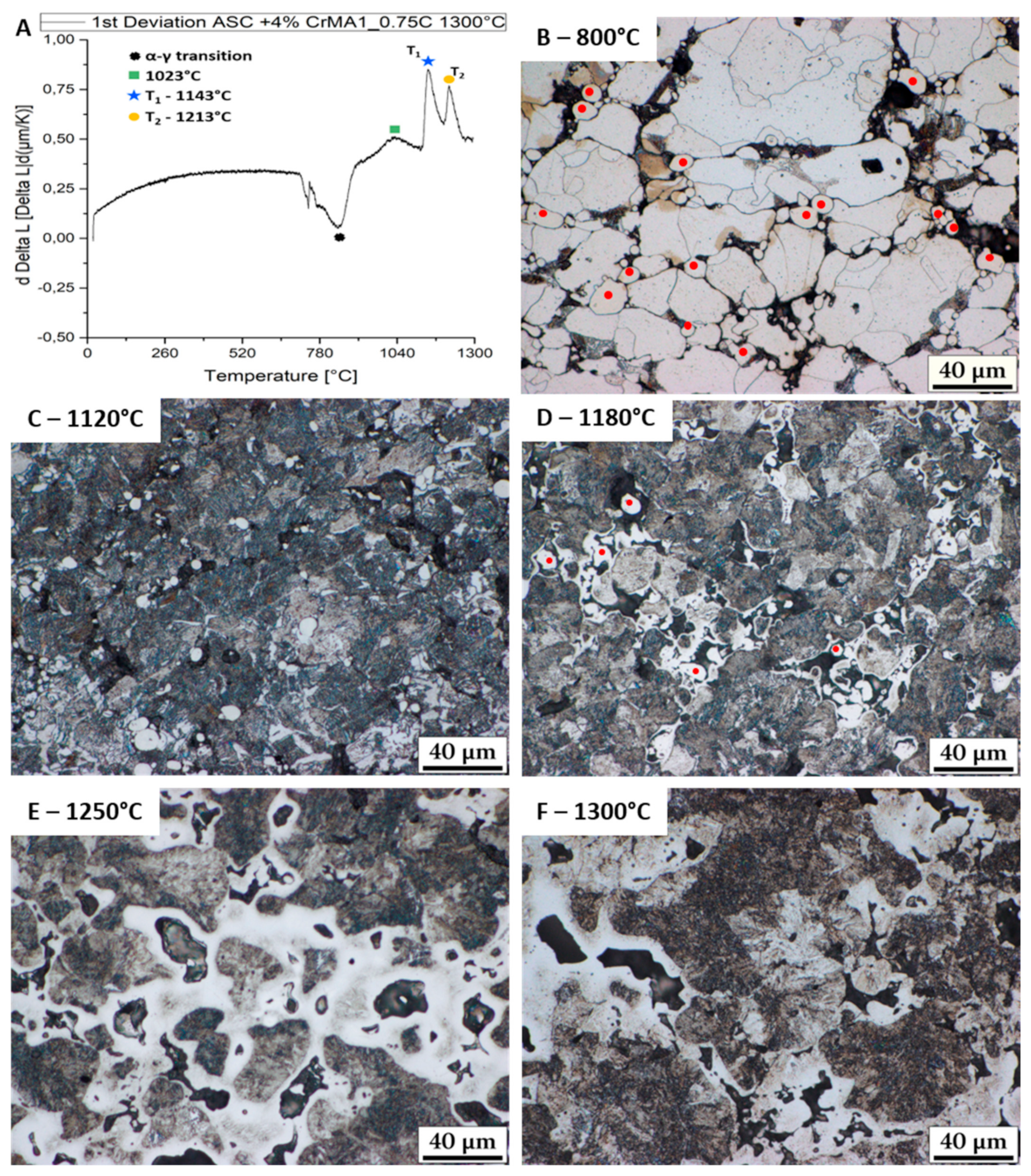
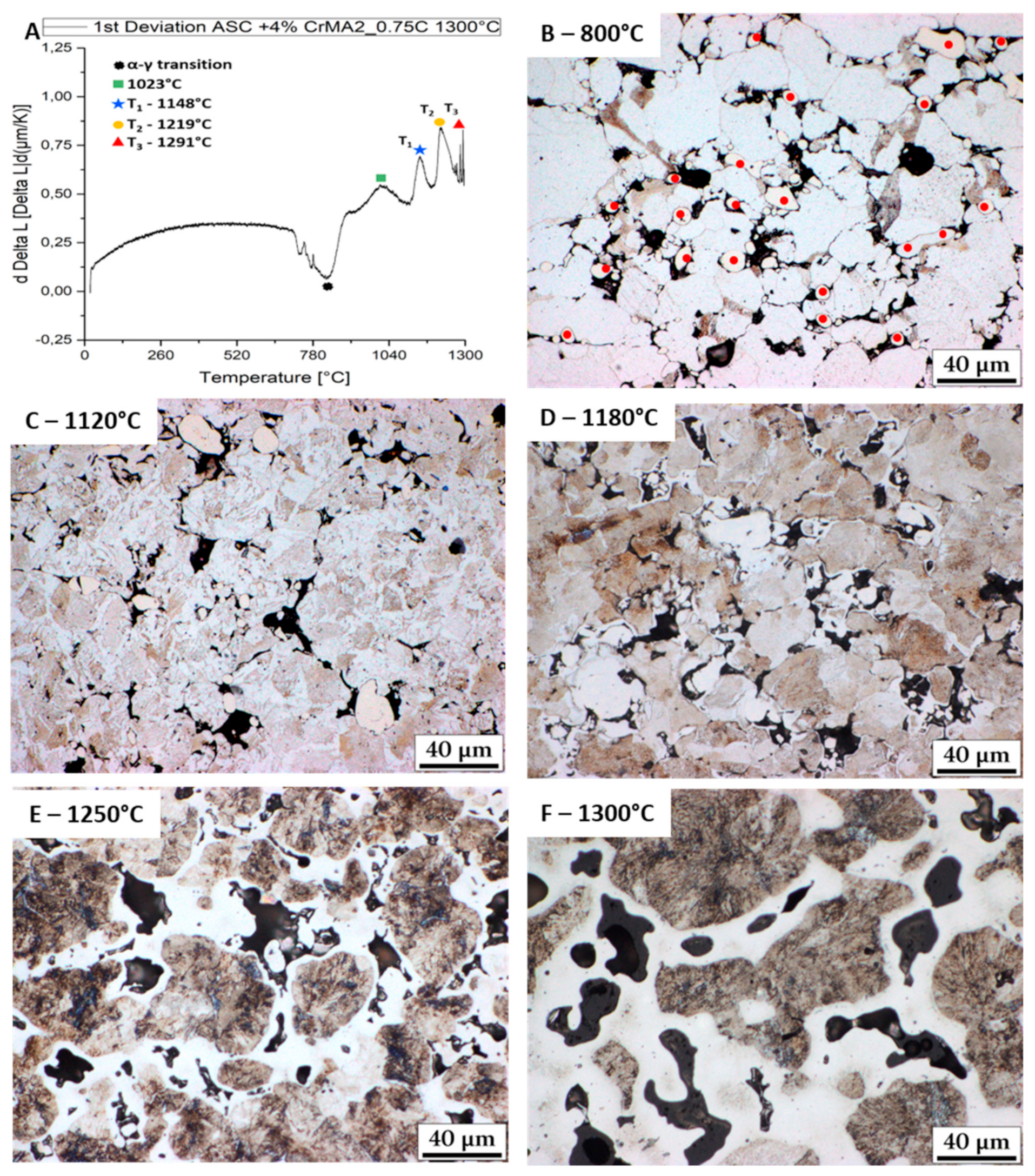
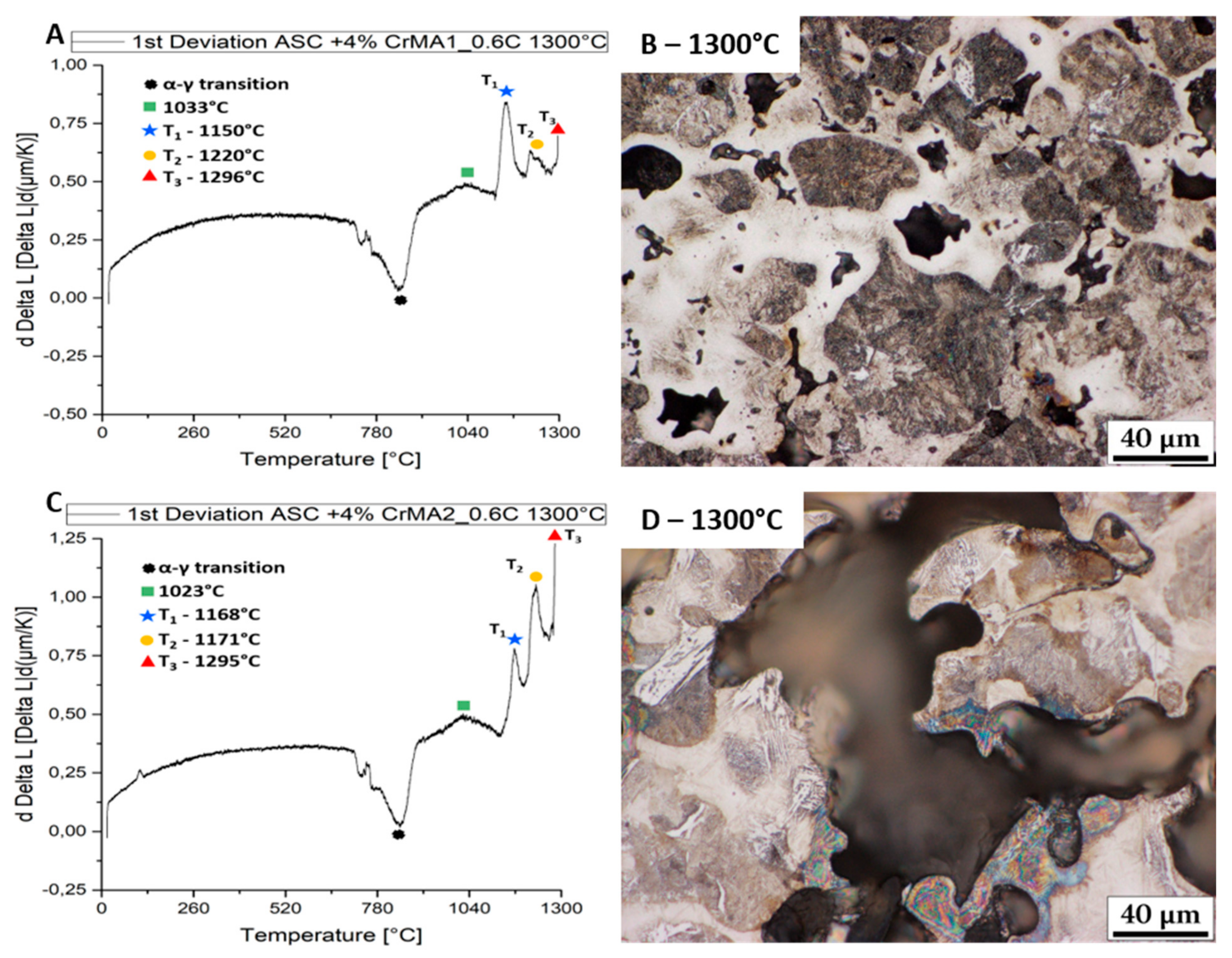
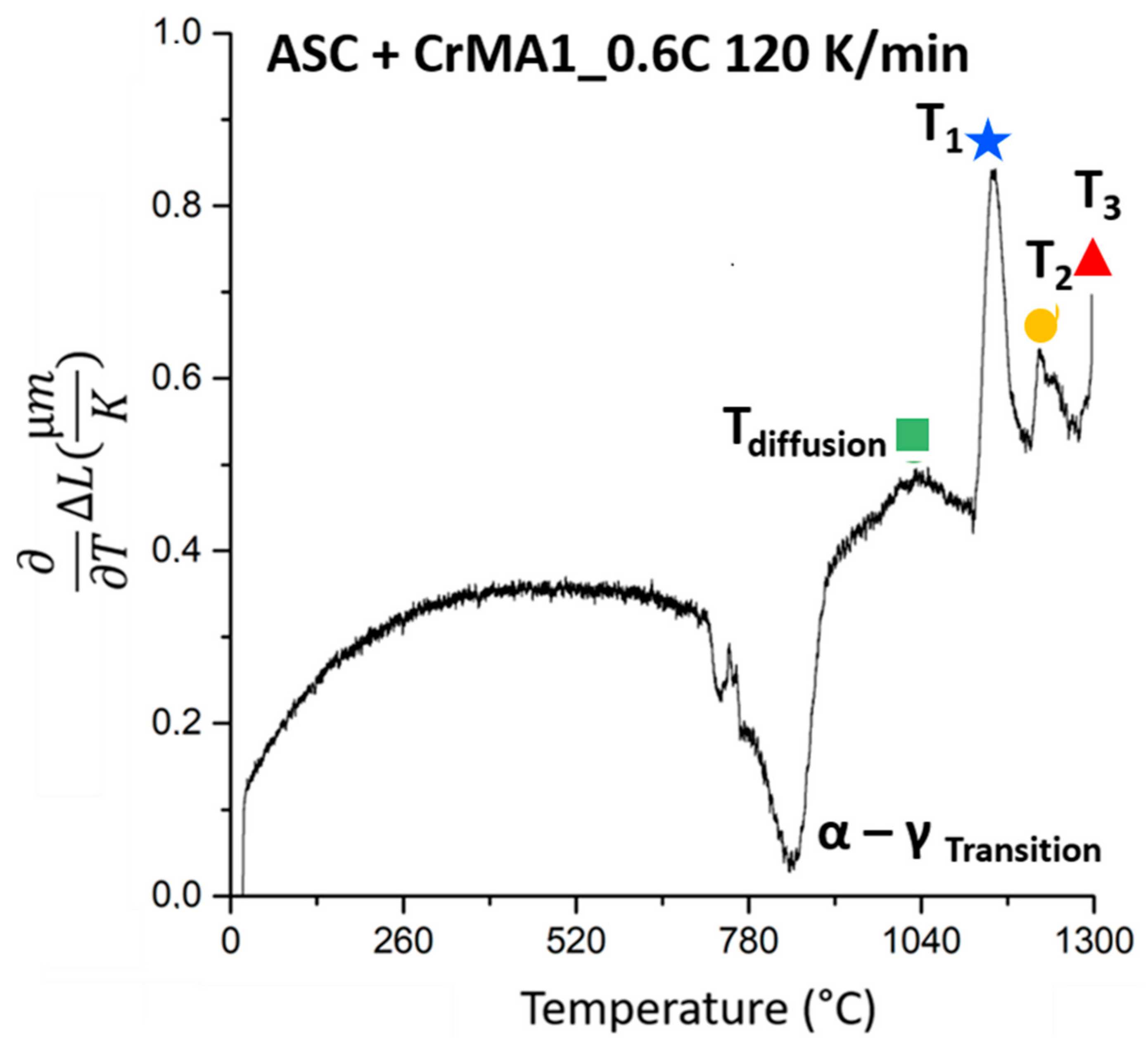
3.1. Evolution of the Melting Process at Fast Heating Rates
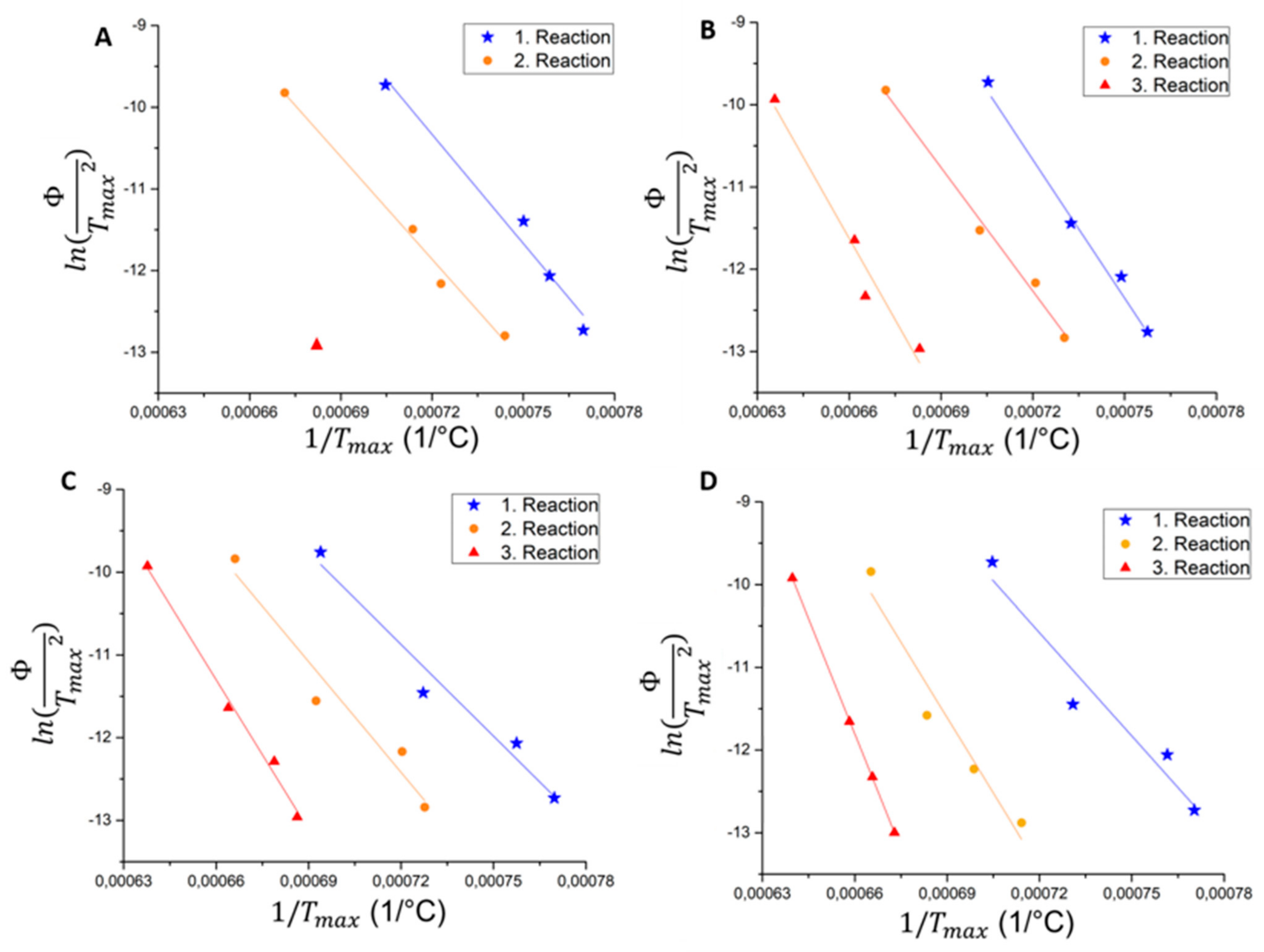
3.2. Influence of the Heating Rate on the Melting Process (5, 10, 20 K/min)
3.3. Hardenability of Samples Sintered at 1180 °C with 10 K/min Heating Rate
4. Conclusions
- Two different materials, plain iron powder ASC + 4% masteralloy CrMA1 and ASC + 4%CrMA2 with two different C-contents each (0.6C) and (0.75C) were studied.
- Quenching dilatometer experiments with fast heating (120 K/min) up to 1300 °C and immediate quenching showed three different melting reactions of the MA’s.
- Further fast heating rate (120 K/min) experiments stopped at different temperatures helped identify the effect of the reactions. The first two melting reactions were considered to be beneficial for the material. The 3rd reaction was identified as causing huge secondary porosity and is to be avoided.
- Slow heating rate (5, 10, 20 K/min) quenching dilatometer experiments were performed up to 1250 °C. These showed that the reaction peak temperatures Tmax are shifted to lower temperatures with lower heating rates.
- Kissinger plots were created from the 1st deviation of dilatometer curves. A linear correlation could be identified for all materials and reactions. This proves that the reaction mechanisms do not change with the heating rate, but the temperatures do. The effect of the heating rate is similar for CrMA1 and CrMA 2.
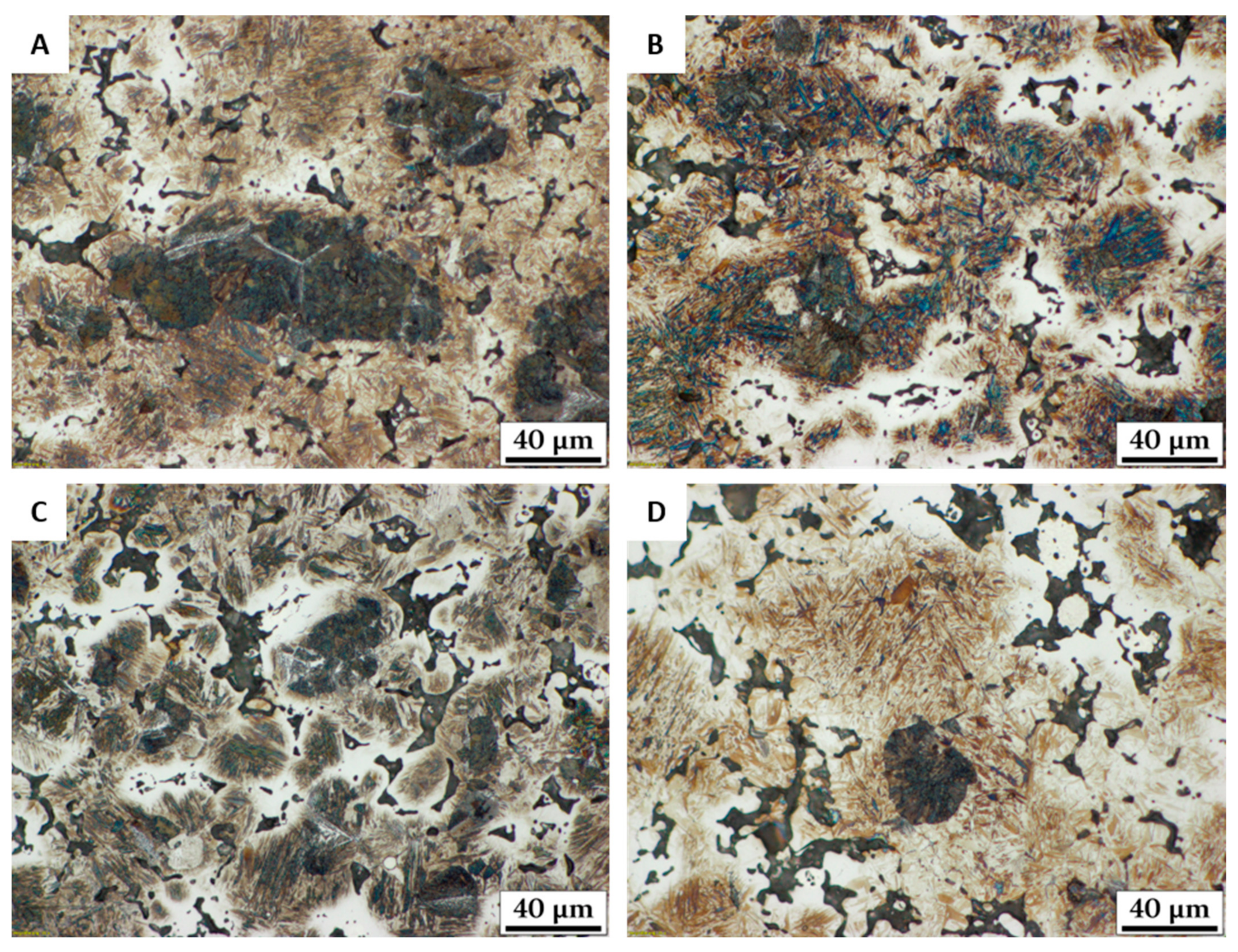
- From these plots 1180 °C was chosen to be a suitable sintering temperature if the material is heated up at 10 K/min. Metallographic sections proved that liquid phase formation and distribution without the formation of secondary porosity could be achieved.
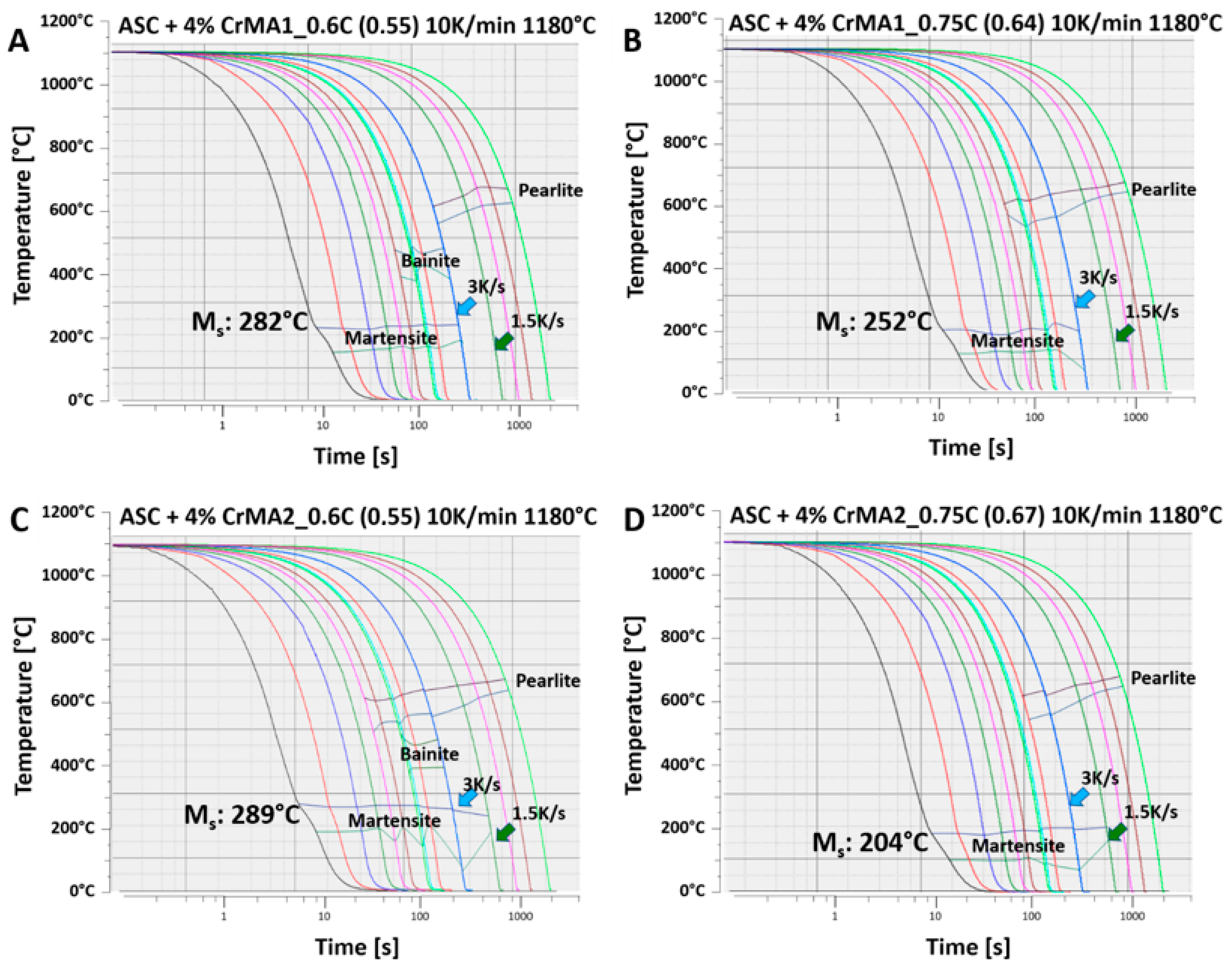
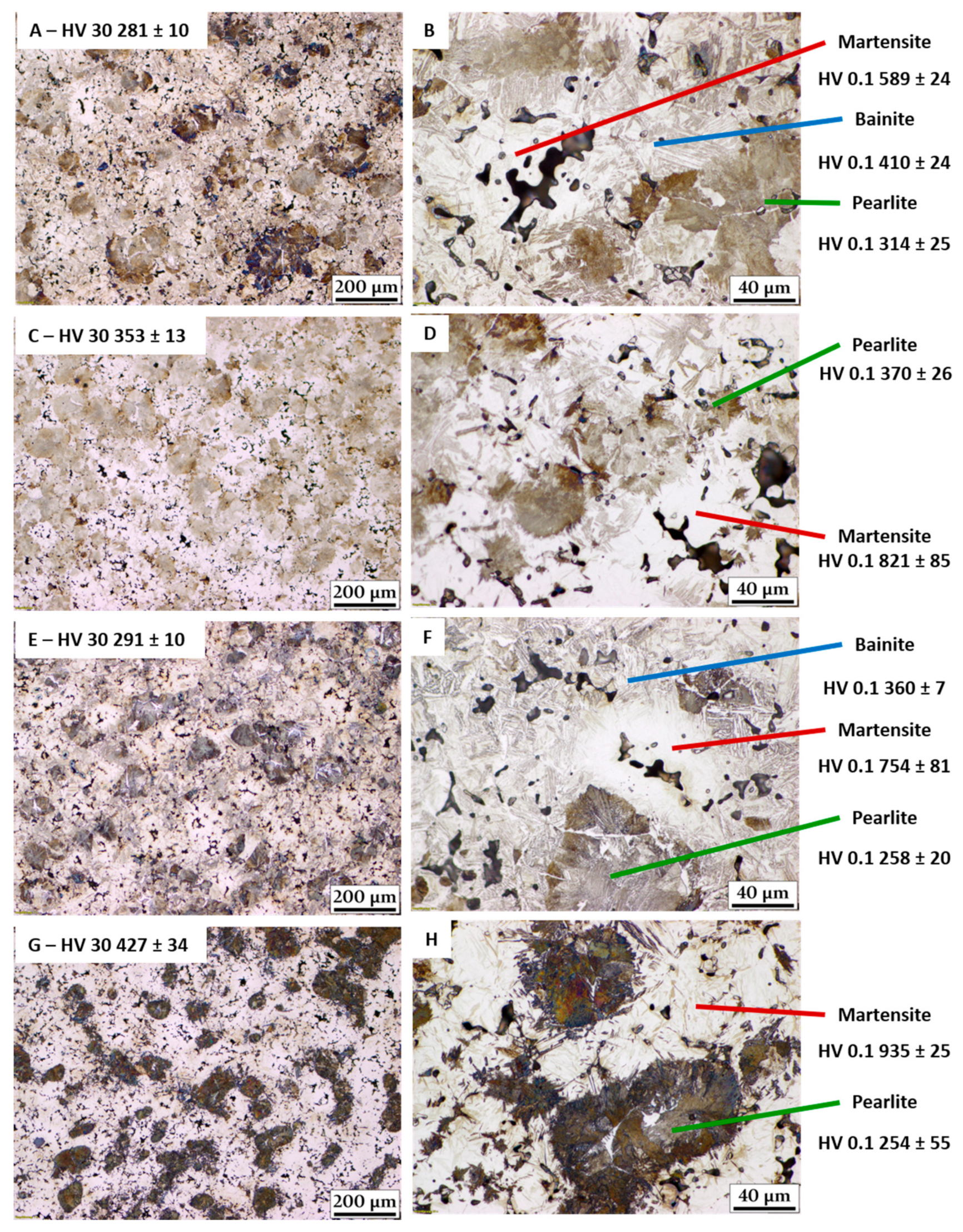
- From the material thus sintered, CCT diagrams were recorded. The phase fields shown in the CCT diagrams could be corroborated with metallographic sections in combination with apparent hardness (HV 30) and microhardness (HV 0.1) measurements.
- Both MAs could melt and distribute the alloying elements as intended via the interconnected porosity. Further homogenization would be a matter of isothermal sintering and therefore diffusion time. There is no disadvantage in using MA containing more alloying elements, as it is designed properly in composition.
- By hot fusion analysis of the O content it could be shown that PM-steel alloyed with Cr via the MA route can be successfully reduced already at 1180 °C.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Copper-Summary. 2021. Available online: https://tradingeconomics.com/commodity/copper (accessed on 4 August 2021).
- Trading Economics. Molybdenum-Summary. 2021. Available online: https://tradingeconomics.com/commodity/molybden (accessed on 4 August 2021).
- European Council. Commission Delegated Regulation (EU) 2015/2420 of 12 October 2015 Amending Council Regulation (EC) No 428/2009 of 5 May 2009. Setting up a Community Regime for the Control of Exports, Transfer, Brokering and Transit of Dual-Use Items, 2015. Off. J. Eur. Union 2015, 616, 295. [Google Scholar]
- Danninger, H.; Gierl-Mayer, C. Advances in powder metallurgy: Properties, processing and applicatons. In Advanced Powder Metallurgy Steel Alloys; Woodhead Publishing: Sawston, UK, 2013; pp. 149–201. [Google Scholar] [CrossRef]
- Lichańska, E.; Sułowski, M.; Ciaś, A. Mechanical properties and microstructure of PM Mn-Cr-Mo steels with low carbon concentration. Arch. Metall. Mater. 2016, 61, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Momeni, M.; Danninger, H.; Gierl, C.; Dudrová, E.; Arvand, A. Relationship between tensile toughness and impact energy of Mo prealloyed and Ni-Cu-Mo diffusion bonded sintered steels. Powder Metall. Prog. 2011, 11, 62–68. [Google Scholar]
- Lindqvist, B. Chormium Alloyed Pm Steels–A New Powder Generation. In Proceedings of the EuroPM2001; European Powder Metallurgy Association (EPMA): Nice, France, 2001; pp. 13–21. [Google Scholar]
- Zapf, G.; Dalal, K. Introduction of high oxygen affinity elements manganese, chromium, and vanadium in the powder metallurgy of P/M parts. Mod. Dev. Powder Metall. 1977, 10, 129–152. [Google Scholar]
- de Oro Calderon, R.; Dunkley, J.; Gierl, C.; Danninger, H. New opportunities for master alloys: Ultra-high pressure water atomised powders. In Powder Metallurgy Review; Inovar Communications Ltd.: Shrewsbury, UK, 2019; pp. 55–66. [Google Scholar]
- Geroldinger, S. Optimization of Iron-Based Master Alloys for Liquid Phase Sintering of PM Steels. Master’s Thesis, Technische Universität Wien, Vienna, Austria, 2019. [Google Scholar]
- Kieback, B.; Schatt, W. Anwendung eines kurzzeitigen Flüssigphasensinterns für die Herstellung von Fe-Ti-Sinterlegierungen. Planseeber. Pulvermetall. 1980, 28, 204–215. [Google Scholar]
- Dautzenberg, N. Dimensional behavior of Copper-Carbon sintered steel. Powder Metall. 1990, 17, 10–15. [Google Scholar]
- Kissinger, H.E. Variation of peak temperature with heating rate in differential thermal analysis. J. Res. Natl. Bur. Stand. 1956, 57, 217–221. [Google Scholar] [CrossRef]
- Kissinger, H.E. Reaction kinetics in differential thermal analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Danninger, H.; Gierl-Mayer, C.; de Oro Calderon, R. The use of transient liquid phases in powder metallurgy. Mater. Sci. For. 2021, 1016, 69–76. [Google Scholar]
- De Oro Calderon, R.; Gierl-Mayer, C.; Danninger, H. Fundamentals of Sintering: Liquid Phase Sintering. Reference Module in Materials Science and Materials Engineering; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar] [CrossRef]
- Danninger, H. Homogenization and pore formation during sintering with transient liquid-phase. Powder Metall. Int. 1988, 20, 21–25. [Google Scholar]
- Danninger, H. Sintering of Mo. alloyed P/M steels prepared from elemental powders. II: Mo. Homogenization and dimensional behaviour. Powder Metall. Int. 1992, 24, 163–168. [Google Scholar]
- Zhang, X. Applications of kinetic methods in thermal analysis: A review. Eng. Sci. 2020, 14, 1–13. [Google Scholar] [CrossRef]
- Wendel, J. Sintering of Water-Atomized Iron and Low-Alloyed Steel Powder. Ph.D. Thesis, Department of Industrial and Materials Science, Chalmers University of Technology, Gothenburg, Sweden, 2020. [Google Scholar]
- Manchili, S.K.; Wendel, J.; Hryha, E.; Nyborg, L. Analysis of iron oxide reduction kinetics in the nanometric scale using hydrogen. Nanomaterials 2020, 10, 1276. [Google Scholar] [CrossRef] [PubMed]
- Sewell, E.C. The consequences for differential thermal analysis of assuming a reaction to be first-order. Clay Miner. 1955, 2, 233–241. [Google Scholar] [CrossRef]
- Oliver, C.; Schuh, C.A. Kissinger-style kinetic analysis for sintering dilatometry data. Metall. Mater. Trans. A 2021, 52, 4479–4487. [Google Scholar] [CrossRef]
- Danninger, H.; Gierl, C. Processes in PM steel compacts during the initial stages of sintering. Mater. Chem. Phys. 2001, 67, 49–55. [Google Scholar] [CrossRef]
- Danninger, H.; Gierl, C.; Kremel, S.; Leitner, G.; Jaenicke-Roessler, K.; Yu, Y. Degassing and deoxidation processes during sintering of unalloyed and alloyed PM steels. Powder Metall. Prog. 2002, 2, 125–140. [Google Scholar]
- Geroldinger, S.; de Oro Calderon, R.; Gierl-Mayer, C.; Danninger, H. Sinter hardening PM steels prepared through hybrid alloying. J. Heat Treat. Mater. 2021, 76, 105–119. [Google Scholar] [CrossRef]
- Steven, W.; Haynes, A.G. The temperature of formation of martensite and bainite in low-alloy steel. J. Iron Steel Inst. 1956, 183, 349–359. [Google Scholar]
- Suzuki, T.; Ono, Y.; Miyamoto, G.; Furuhara, T. Effects of Si and Cr on bainite microstructure of medium carbon steels. Iron Steel Int. J. 2010, 50, 1476–1482. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Material | |||
|---|---|---|---|---|
| C | Natural Graphite UF 4 | |||
| Fe | Base powder: | Plain iron, ASC 100.29 | d50: 52.38 ± 3.00 µm | d90: 75.89 ± 6.81 µm |
| CrMA-1 | MA: | Fe_32Cr_8Si_4C | d50: 6.80 ± 0.19 µm | d90: 16.76 ± 0.58 µm |
| CrMA-2 | MA: | Fe_40Cr_11Si_3.5C | d50: 8.10 ± 0.86 µm | d90: 20.16 ± 2.38 µm |
| Material | C [wt%] | O [wt%] | Material | C [wt%] | O [wt%] |
|---|---|---|---|---|---|
| ASC + CrMA1_0.6C 800 °C p.s. | 0.595 ± 0.007 | 0..073 ± 0.005 | ASC + CrMA1_0.6C 1180 °C | 0.554 ± 0.003 | 0.020 ± 0.001 |
| ASC + CrMA1_0.75C 800 °C p.s. | 0..725 ± 0.013 | 0.073± 0.002 | ASC + CrMA1_0.75C 1180 °C | 0.636 ± 0.024 | 0.018 ± 0.001 |
| ASC + CrMA2_0.6C 800 °C p.s. | 0.593 ± 0.004 | 0.072 ± 0.002 | ASC + CrMA2_0.6C 1180 °C | 0.539 ± 0.015 | 0.022 ± 0.001 |
| ASC + CrMA2_0.75C 800 °C p.s. | 0.723 ± 0.008 | 0.076 ± 0.006 | ASC + CrMA2_0.75C 1180 °C | 0.667 ± 0.012 | 0.022 ± 0.001 |
| Heating Rate | ASC + 4%CrMA1_0.6C | Heating Rate | ASC + 4%CrMA1_0.75C | ||||
|---|---|---|---|---|---|---|---|
| (K/min) | T1 (°C) | T2 (°C) | T3 (°C) | (K/min) | T1 (°C) | T2 (°C) | T3 (°C) |
| 5 | 1026 | 1071 | 1223 | 5 | 1047 | 1096 | 1191 |
| 10 | 1045 | 1110 | 1159 | 10 | 1062 | 1114 | x |
| 20 | 1060 | 1128 | 1187 | 20 | 1092 | 1150 | x |
| 120 | 1146 | 1216 | 1289 | 120 | 1145 | 1215 | x |
| Heating Rate | ASC + 4%CrMA2_0.6C | Heating Rate | ASC + 4%CrMA2_0.75C | ||||
| (K/min) | T1 (°C) | T2 (°C) | T3 (°C) | (K/min) | T1 (°C) | T2 (°C) | T3 (°C) |
| 5 | 1026 | 1101 | 1184 | 5 | 1025 | 1127 | 1213 |
| 10 | 1047 | 1115 | 1200 | 10 | 1040 | 1158 | 1229 |
| 20 | 1102 | 1171 | 1233 | 20 | 1095 | 1190 | 1246 |
| 120 | 1168 | 1228 | 1275 | 120 | 1146 | 1235 | 1290 |
| Material | Ms Measured [°C] | Ms Calculated | ΔT [°C] |
| ASC + 4% CrMA1_0.6C (0.55C) | 282 | 275 | −6 |
| ASC + 4% CrMA1_0.75C (0.64C) | 252 | 233 | −19 |
| ASC + 4% CrMA2_0.6C (0.55C) | 289 | 273 | −16 |
| ASC + 4% CrMA2_0.75C (0.64C) | 204 | 212 | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geroldinger, S.; de Oro Calderon, R.; Gierl-Mayer, C.; Danninger, H. Transient Liquid Phase Sintering of PM Steel—A Matter of the Heating Rate. Metals 2021, 11, 1662. https://doi.org/10.3390/met11101662
Geroldinger S, de Oro Calderon R, Gierl-Mayer C, Danninger H. Transient Liquid Phase Sintering of PM Steel—A Matter of the Heating Rate. Metals. 2021; 11(10):1662. https://doi.org/10.3390/met11101662
Chicago/Turabian StyleGeroldinger, Stefan, Raquel de Oro Calderon, Christian Gierl-Mayer, and Herbert Danninger. 2021. "Transient Liquid Phase Sintering of PM Steel—A Matter of the Heating Rate" Metals 11, no. 10: 1662. https://doi.org/10.3390/met11101662