Targeting SWI/SNF Complexes in Cancer: Pharmacological Approaches and Implications



Abstract
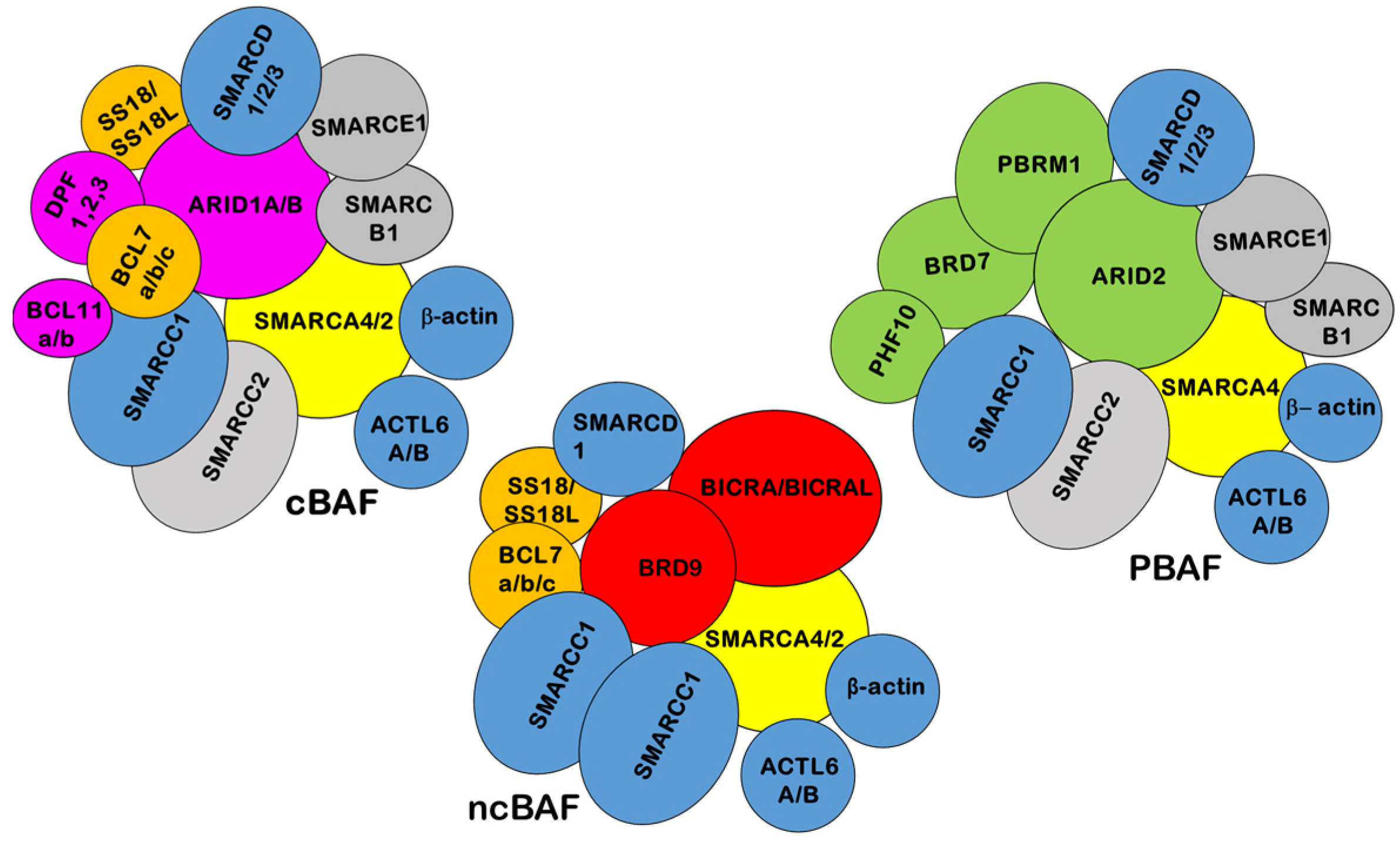
:1. SWI/SNF Structure and Function
2. SWI/SNF in Cancer
3. Pharmacological Inhibition of SWI/SNF Function
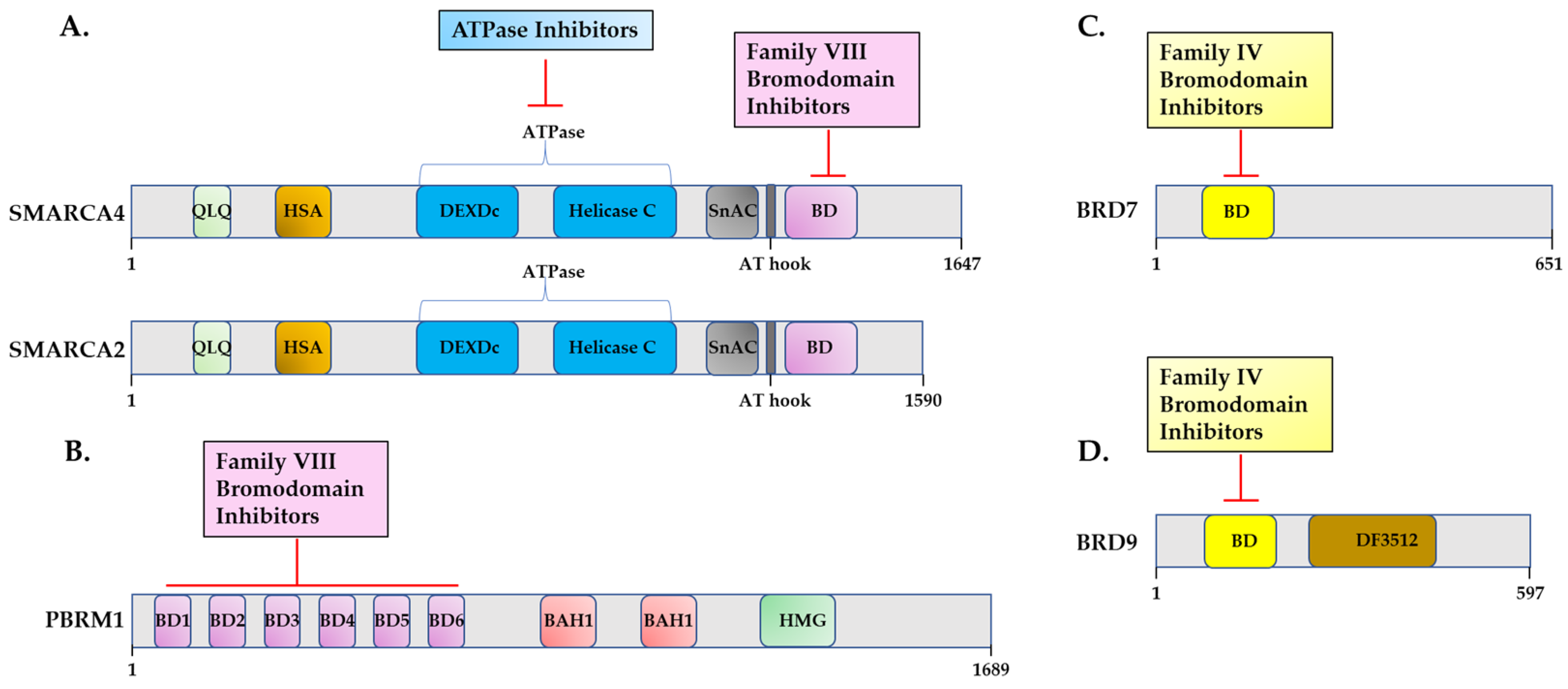
3.1. Inhibitors of SWI/SNF Catalytic Activity
3.2. Targeting SWI/SNF Bromodomains
3.2.1. Targeting SWI/SNF Family VIII Bromodomains
3.2.2. Targeting SWI/SNF Family IV Bromodomains
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hernandez-Garcia, J.; Diego-Martin, B.; Kuo, P.H.; Jami-Alahmadi, Y.; Vashisht, A.A.; Wohlschlegel, J.; Jacobsen, S.E.; Blazquez, M.A.; Gallego-Bartolome, J. Comprehensive identification of SWI/SNF complex subunits underpins deep eukaryotic ancestry and reveals new plant components. Commun. Biol. 2022, 5, 549. [Google Scholar] [CrossRef]
- Centore, R.C.; Sandoval, G.J.; Soares, L.M.M.; Kadoch, C.; Chan, H.M. Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet. 2020, 36, 936–950. [Google Scholar] [CrossRef]
- Yan, Z.; Cui, K.; Murray, D.M.; Ling, C.; Xue, Y.; Gerstein, A.; Parsons, R.; Zhao, K.; Wang, W. PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev. 2005, 19, 1662–1667. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Canman, J.C.; Lee, C.S.; Nie, Z.; Yang, D.; Moreno, G.T.; Young, M.K.; Salmon, E.D.; Wang, W. The human SWI/SNF-B chromatin-remodeling complex is related to yeast rsc and localizes at kinetochores of mitotic chromosomes. Proc. Natl. Acad. Sci. USA 2000, 97, 13015–13020. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.C.; D’Avino, A.R.; Cassel, S.H.; Mashtalir, N.; McKenzie, Z.M.; McBride, M.J.; Valencia, A.M.; Zhou, Q.; Bocker, M.; Soares, L.M.M.; et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 2018, 20, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Mashtalir, N.; D’Avino, A.R.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; St Pierre, R.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288.e20. [Google Scholar] [CrossRef] [PubMed]
- Alpsoy, A.; Dykhuizen, E.C. Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. J. Biol. Chem. 2018, 293, 3892–3903. [Google Scholar] [CrossRef] [PubMed]
- Gatchalian, J.; Malik, S.; Ho, J.; Lee, D.S.; Kelso, T.W.R.; Shokhirev, M.N.; Dixon, J.R.; Hargreaves, D.C. A non-canonical BRD9-containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat. Commun. 2018, 9, 5139. [Google Scholar] [CrossRef]
- Wang, X.; Wang, S.; Troisi, E.C.; Howard, T.P.; Haswell, J.R.; Wolf, B.K.; Hawk, W.H.; Ramos, P.; Oberlick, E.M.; Tzvetkov, E.P.; et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun. 2019, 10, 1881. [Google Scholar] [CrossRef]
- Olave, I.; Wang, W.; Xue, Y.; Kuo, A.; Crabtree, G.R. Identification of a polymorphic, neuron-specific chromatin remodeling complex. Genes Dev. 2002, 16, 2509–2517. [Google Scholar] [CrossRef]
- Soshnikova, N.V.; Azieva, A.M.; Klimenko, N.S.; Khamidullina, A.I.; Feoktistov, A.V.; Sheynov, A.A.; Brechalov, A.V.; Tatarskiy, V.V.; Georgieva, S.G. A novel chromatin-remodeling complex variant, dcPBAF, is involved in maintaining transcription in differentiated neurons. Front. Cell Dev. Biol. 2023, 11, 1271598. [Google Scholar] [CrossRef] [PubMed]
- Imbalzano, A.N.; Kwon, H.; Green, M.R.; Kingston, R.E. Facilitated binding of TATA-binding protein to nucleosomal DNA. Nature 1994, 370, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Imbalzano, A.N.; Khavari, P.A.; Kingston, R.E.; Green, M.R. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature 1994, 370, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Narlikar, G.J.; Phelan, M.L.; Kingston, R.E. Generation and interconversion of multiple distinct nucleosomal states as a mechanism for catalyzing chromatin fluidity. Mol. Cell 2001, 8, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Dechassa, M.L.; Sabri, A.; Pondugula, S.; Kassabov, S.R.; Chatterjee, N.; Kladde, M.P.; Bartholomew, B. SWI/SNF has intrinsic nucleosome disassembly activity that is dependent on adjacent nucleosomes. Mol. Cell 2010, 38, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Hada, A.; Sen, P.; Olufemi, L.; Hall, M.A.; Smith, B.Y.; Forth, S.; McKnight, J.N.; Patel, A.; Bowman, G.D.; et al. Dynamic regulation of transcription factors by nucleosome remodeling. eLife 2015, 4, e06249. [Google Scholar] [CrossRef]
- Phelan, M.L.; Sif, S.; Narlikar, G.J.; Kingston, R.E. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol. Cell 1999, 3, 247–253. [Google Scholar] [CrossRef]
- Valencia, A.M.; Collings, C.K.; Dao, H.T.; St Pierre, R.; Cheng, Y.C.; Huang, J.; Sun, Z.Y.; Seo, H.S.; Mashtalir, N.; Comstock, D.E.; et al. Recurrent SMARCB1 Mutations Reveal a Nucleosome Acidic Patch Interaction Site That Potentiates mSWI/SNF Complex Chromatin Remodeling. Cell 2019, 179, 1342–1356.e23. [Google Scholar] [CrossRef]
- Han, Y.; Reyes, A.A.; Malik, S.; He, Y. Cryo-EM structure of SWI/SNF complex bound to a nucleosome. Nature 2020, 579, 452–455. [Google Scholar] [CrossRef]
- He, S.; Wu, Z.; Tian, Y.; Yu, Z.; Yu, J.; Wang, X.; Li, J.; Liu, B.; Xu, Y. Structure of nucleosome-bound human BAF complex. Science 2020, 367, 875–881. [Google Scholar] [CrossRef]
- Chen, J.; Archer, T.K. Regulating SWI/SNF subunit levels via protein-protein interactions and proteasomal degradation: BAF155 and BAF170 limit expression of BAF57. Mol. Cell. Biol. 2005, 25, 9016–9027. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, K.; Zhang, W.; Chen, Z. Structure of human chromatin-remodelling PBAF complex bound to a nucleosome. Nature 2022, 605, 166–171. [Google Scholar] [CrossRef]
- Wang, L.; Yu, J.; Yu, Z.; Wang, Q.; Li, W.; Ren, Y.; Chen, Z.; He, S.; Xu, Y. Structure of nucleosome-bound human PBAF complex. Nat. Commun. 2022, 13, 7644. [Google Scholar] [CrossRef]
- Schubert, H.L.; Wittmeyer, J.; Kasten, M.M.; Hinata, K.; Rawling, D.C.; Heroux, A.; Cairns, B.R.; Hill, C.P. Structure of an actin-related subcomplex of the SWI/SNF chromatin remodeler. Proc. Natl. Acad. Sci. USA 2013, 110, 3345–3350. [Google Scholar] [CrossRef]
- Clapier, C.R.; Verma, N.; Parnell, T.J.; Cairns, B.R. Cancer-Associated Gain-of-Function Mutations Activate a SWI/SNF-Family Regulatory Hub. Mol. Cell 2020, 80, 712–725.e5. [Google Scholar] [CrossRef] [PubMed]
- Morrison, E.A.; Sanchez, J.C.; Ronan, J.L.; Farrell, D.P.; Varzavand, K.; Johnson, J.K.; Gu, B.X.; Crabtree, G.R.; Musselman, C.A. DNA binding drives the association of BRG1/hBRM bromodomains with nucleosomes. Nat. Commun. 2017, 8, 16080. [Google Scholar] [CrossRef] [PubMed]
- Porter, E.G.; Dykhuizen, E.C. Individual Bromodomains of Polybromo-1 Contribute to Chromatin Association and Tumor Suppression in Clear Cell Renal Carcinoma. J. Biol. Chem. 2017, 292, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zhou, J.; Liu, H.Y.; Zhou, M.; Wang, L.L.; Zhang, Q.H.; Yang, Y.X.; Xiong, W.; Shen, S.R.; Li, X.L.; et al. The transcriptional regulation role of BRD7 by binding to acetylated histone through bromodomain. J. Cell. Biochem. 2006, 97, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Flynn, E.M.; Huang, O.W.; Poy, F.; Oppikofer, M.; Bellon, S.F.; Tang, Y.; Cochran, A.G. A Subset of Human Bromodomains Recognizes Butyryllysine and Crotonyllysine Histone Peptide Modifications. Structure 2015, 23, 1801–1814. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Yamauchi, M.; Nishina, M.; Yamamichi, N.; Mizutani, T.; Ui, M.; Murakami, M.; Iba, H. Identification of SWI.SNF complex subunit BAF60a as a determinant of the transactivation potential of Fos/Jun dimers. J. Biol. Chem. 2001, 276, 2852–2857. [Google Scholar] [CrossRef] [PubMed]
- Simone, C.; Forcales, S.V.; Hill, D.A.; Imbalzano, A.N.; Latella, L.; Puri, P.L. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat. Genet. 2004, 36, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Link, K.A.; Burd, C.J.; Williams, E.; Marshall, T.; Rosson, G.; Henry, E.; Weissman, B.; Knudsen, K.E. BAF57 governs androgen receptor action and androgen-dependent proliferation through SWI/SNF. Mol. Cell. Biol. 2005, 25, 2200–2215. [Google Scholar] [CrossRef]
- Priam, P.; Krasteva, V.; Rousseau, P.; D’Angelo, G.; Gaboury, L.; Sauvageau, G.; Lessard, J.A. SMARCD2 subunit of SWI/SNF chromatin-remodeling complexes mediates granulopoiesis through a CEBPvarepsilon dependent mechanism. Nat. Genet. 2017, 49, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Witzel, M.; Petersheim, D.; Fan, Y.; Bahrami, E.; Racek, T.; Rohlfs, M.; Puchalka, J.; Mertes, C.; Gagneur, J.; Ziegenhain, C.; et al. Chromatin-remodeling factor SMARCD2 regulates transcriptional networks controlling differentiation of neutrophil granulocytes. Nat. Genet. 2017, 49, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Aras, S.; Saladi, S.V.; Basuroy, T.; Marathe, H.G.; Lores, P.; de la Serna, I.L. BAF60A mediates interactions between the microphthalmia-associated transcription factor and the BRG1-containing SWI/SNF complex during melanocyte differentiation. J. Cell. Physiol. 2019, 234, 11780–11791. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Sohn, D.H.; Ko, M.; Chung, H.; Jeon, S.H.; Seong, R.H. BAF60a interacts with p53 to recruit the SWI/SNF complex. J. Biol. Chem. 2008, 283, 11924–11934. [Google Scholar] [CrossRef]
- Hu, X.; Liu, R.; Hou, J.; Peng, W.; Wan, S.; Xu, M.; Li, Y.; Zhang, G.; Zhai, X.; Liang, P.; et al. SMARCE1 promotes neuroblastoma tumorigenesis through assisting MYCN-mediated transcriptional activation. Oncogene 2022, 41, 4295–4306. [Google Scholar] [CrossRef]
- Feng, J.; Xu, X.; Fan, X.; Yi, Q.; Tang, L. BAF57/SMARCE1 Interacting with Splicing Factor SRSF1 Regulates Mechanical Stress-Induced Alternative Splicing of Cyclin D1. Genes 2021, 12, 306. [Google Scholar] [CrossRef]
- Singh, A.P.; Archer, T.K. Analysis of the SWI/SNF chromatin-remodeling complex during early heart development and BAF250a repression cardiac gene transcription during P19 cell differentiation. Nucleic Acids Res. 2014, 42, 2958–2975. [Google Scholar] [CrossRef]
- Wang, X.; Song, C.; Ye, Y.; Gu, Y.; Li, X.; Chen, P.; Leng, D.; Xiao, J.; Wu, H.; Xie, S.; et al. BRD9-mediated control of the TGF-beta/Activin/Nodal pathway regulates self-renewal and differentiation of human embryonic stem cells and progression of cancer cells. Nucleic Acids Res. 2023, 51, 11634–11651. [Google Scholar] [CrossRef]
- Chandler, R.L.; Magnuson, T. The SWI/SNF BAF-A complex is essential for neural crest development. Dev. Biol. 2016, 411, 15–24. [Google Scholar] [CrossRef]
- Menon, D.U.; Kirsanov, O.; Geyer, C.B.; Magnuson, T. Mammalian SWI/SNF chromatin remodeler is essential for reductional meiosis in males. Nat. Commun. 2021, 12, 6581. [Google Scholar] [CrossRef]
- Yamashita, N.; Morimoto, Y.; Fushimi, A.; Ahmad, R.; Bhattacharya, A.; Daimon, T.; Haratake, N.; Inoue, Y.; Ishikawa, S.; Yamamoto, M.; et al. MUC1-C Dictates PBRM1-Mediated Chronic Induction of Interferon Signaling, DNA Damage Resistance, and Immunosuppression in Triple-Negative Breast Cancer. Mol. Cancer Res. 2023, 21, 274–289. [Google Scholar] [CrossRef]
- Padilla-Benavides, T.; Olea-Flores, M.; Sharma, T.; Syed, S.A.; Witwicka, H.; Zuniga-Eulogio, M.D.; Zhang, K.; Navarro-Tito, N.; Imbalzano, A.N. Differential Contributions of mSWI/SNF Chromatin Remodeler Sub-Families to Myoblast Differentiation. Int. J. Mol. Sci. 2023, 24, 11256. [Google Scholar] [CrossRef] [PubMed]
- Alpsoy, A.; Utturkar, S.M.; Carter, B.C.; Dhiman, A.; Torregrosa-Allen, S.E.; Currie, M.P.; Elzey, B.D.; Dykhuizen, E.C. BRD9 Is a Critical Regulator of Androgen Receptor Signaling and Prostate Cancer Progression. Cancer Res. 2021, 81, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, J.; Chen, X. The activity of p53 is differentially regulated by Brm- and Brg1-containing SWI/SNF chromatin remodeling complexes. J. Biol. Chem. 2007, 282, 37429–37435. [Google Scholar] [CrossRef] [PubMed]
- Reisman, D.N.; Strobeck, M.W.; Betz, B.L.; Sciariotta, J.; Funkhouser, W., Jr.; Murchardt, C.; Yaniv, M.; Sherman, L.S.; Knudsen, E.S.; Weissman, B.E. Concomitant down-regulation of BRM and BRG1 in human tumor cell lines: Differential effects on RB-mediated growth arrest vs CD44 expression. Oncogene 2002, 21, 1196–1207. [Google Scholar] [CrossRef]
- Kelso, T.W.R.; Porter, D.K.; Amaral, M.L.; Shokhirev, M.N.; Benner, C.; Hargreaves, D.C. Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. eLife 2017, 6, e30506. [Google Scholar] [CrossRef]
- Baxter, A.E.; Huang, H.; Giles, J.R.; Chen, Z.; Wu, J.E.; Drury, S.; Dalton, K.; Park, S.L.; Torres, L.; Simone, B.W.; et al. The SWI/SNF chromatin remodeling complexes BAF and PBAF differentially regulate epigenetic transitions in exhausted CD8(+) T cells. Immunity 2023, 56, 1320–1340.e10. [Google Scholar] [CrossRef]
- Kharel, A.; Shen, J.; Brown, R.; Chen, Y.; Nguyen, C.; Alson, D.; Bluemn, T.; Fan, J.; Gai, K.; Zhang, B.; et al. Loss of PBAF promotes expansion and effector differentiation of CD8(+) T cells during chronic viral infection and cancer. Cell Rep. 2023, 42, 112649. [Google Scholar] [CrossRef]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bosse, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef]
- Pan, D.; Kobayashi, A.; Jiang, P.; Ferrari de Andrade, L.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef]
- Mathur, R.; Alver, B.H.; San Roman, A.K.; Wilson, B.G.; Wang, X.; Agoston, A.T.; Park, P.J.; Shivdasani, R.A.; Roberts, C.W. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 2017, 49, 296–302. [Google Scholar] [CrossRef]
- McDonald, B.; Chick, B.Y.; Ahmed, N.S.; Burns, M.; Ma, S.; Casillas, E.; Chen, D.; Mann, T.H.; O’Connor, C.; Hah, N.; et al. Canonical BAF complex activity shapes the enhancer landscape that licenses CD8(+) T cell effector and memory fates. Immunity 2023, 56, 1303–1319.e5. [Google Scholar] [CrossRef]
- Carcamo, S.; Nguyen, C.B.; Grossi, E.; Filipescu, D.; Alpsoy, A.; Dhiman, A.; Sun, D.; Narang, S.; Imig, J.; Martin, T.C.; et al. Altered BAF occupancy and transcription factor dynamics in PBAF-deficient melanoma. Cell Rep. 2022, 39, 110637. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, K.; Jia, Y.; Chuang, J.C.; Sun, X.; Lin, Y.H.; Celen, C.; Li, L.; Huang, F.; Liu, X.; et al. Dual ARID1A/ARID1B loss leads to rapid carcinogenesis and disruptive redistribution of BAF complexes. Nat. Cancer 2020, 1, 909–922. [Google Scholar] [CrossRef] [PubMed]
- St Pierre, R.; Collings, C.K.; Same Guerra, D.D.; Widmer, C.J.; Bolonduro, O.; Mashtalir, N.; Sankar, A.; Liang, Y.; Bi, W.L.; Gerkes, E.H.; et al. SMARCE1 deficiency generates a targetable mSWI/SNF dependency in clear cell meningioma. Nat. Genet. 2022, 54, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Brahma, S.; Henikoff, S. Epigenetic pioneering by SWI/SNF family remodelers. Mol. Cell 2024, 84, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Schick, S.; Grosche, S.; Kohl, K.E.; Drpic, D.; Jaeger, M.G.; Marella, N.C.; Imrichova, H.; Lin, J.G.; Hofstatter, G.; Schuster, M.; et al. Acute BAF perturbation causes immediate changes in chromatin accessibility. Nat. Genet. 2021, 53, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, M.; Stadler, M.B.; Masoni, F.; Jagani, Z.; Galli, G.G.; Schubeler, D. Mammalian SWI/SNF continuously restores local accessibility to chromatin. Nat. Genet. 2021, 53, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, D.C. Chromatin openness requires continuous SWI/SNF activity. Nat. Genet. 2021, 53, 263–264. [Google Scholar] [CrossRef]
- Davo-Martinez, C.; Helfricht, A.; Ribeiro-Silva, C.; Raams, A.; Tresini, M.; Uruci, S.; van Cappellen, W.A.; Taneja, N.; Demmers, J.A.A.; Pines, A.; et al. Different SWI/SNF complexes coordinately promote R-loop- and RAD52-dependent transcription-coupled homologous recombination. Nucleic Acids Res. 2023, 51, 9055–9074. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Huang, J.; Zhang, C.; Zhao, F.; Kim, W.; Tu, X.; Zhang, Y.; Nowsheen, S.; Zhu, Q.; Deng, M.; et al. The bromodomain containing protein BRD-9 orchestrates RAD51-RAD54 complex formation and regulates homologous recombination-mediated repair. Nat. Commun. 2020, 11, 2639. [Google Scholar] [CrossRef] [PubMed]
- Kakarougkas, A.; Ismail, A.; Chambers, A.L.; Riballo, E.; Herbert, A.D.; Kunzel, J.; Lobrich, M.; Jeggo, P.A.; Downs, J.A. Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol. Cell 2014, 55, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Ui, A.; Kanno, S.; Ogiwara, H.; Nagase, T.; Kohno, T.; Yasui, A. SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res. 2014, 74, 2465–2475. [Google Scholar] [CrossRef]
- Lans, H.; Marteijn, J.A.; Schumacher, B.; Hoeijmakers, J.H.; Jansen, G.; Vermeulen, W. Involvement of global genome repair, transcription coupled repair, and chromatin remodeling in UV DNA damage response changes during development. PLoS Genet. 2010, 6, e1000941. [Google Scholar] [CrossRef] [PubMed]
- Hara, R.; Sancar, A. The SWI/SNF chromatin-remodeling factor stimulates repair by human excision nuclease in the mononucleosome core particle. Mol. Cell. Biol. 2002, 22, 6779–6787. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Fitzgerald, D.J.; Smith, C.L.; Peterson, C.L.; Richmond, T.J.; Thoma, F. Chromatin remodeling activities act on UV-damaged nucleosomes and modulate DNA damage accessibility to photolyase. J. Biol. Chem. 2003, 278, 17655–17663. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Q.; Jones, K.; Patel, M.; Gong, F. The chromatin remodeling factor BRG1 stimulates nucleotide excision repair by facilitating recruitment of XPC to sites of DNA damage. Cell Cycle 2009, 8, 3953–3959. [Google Scholar] [CrossRef]
- Ray, A.; Mir, S.N.; Wani, G.; Zhao, Q.; Battu, A.; Zhu, Q.; Wang, Q.E.; Wani, A.A. Human SNF5/INI1, a component of the human SWI/SNF chromatin remodeling complex, promotes nucleotide excision repair by influencing ATM recruitment and downstream H2AX phosphorylation. Mol. Cell. Biol. 2009, 29, 6206–6219. [Google Scholar] [CrossRef]
- Kothandapani, A.; Gopalakrishnan, K.; Kahali, B.; Reisman, D.; Patrick, S.M. Downregulation of SWI/SNF chromatin remodeling factor subunits modulates cisplatin cytotoxicity. Exp. Cell Res. 2012, 318, 1973–1986. [Google Scholar] [CrossRef]
- Menoni, H.; Gasparutto, D.; Hamiche, A.; Cadet, J.; Dimitrov, S.; Bouvet, P.; Angelov, D. ATP-dependent chromatin remodeling is required for base excision repair in conventional but not in variant H2A.Bbd nucleosomes. Mol. Cell. Biol. 2007, 27, 5949–5956. [Google Scholar] [CrossRef]
- Yu, Z.C.; Li, T.; Tully, E.; Huang, P.; Chen, C.N.; Oberdoerffer, P.; Gaillard, S.; Shih, I.M.; Wang, T.L. Temozolomide Sensitizes ARID1A-Mutated Cancers to PARP Inhibitors. Cancer Res. 2023, 83, 2750–2762. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Jung, H.Y.; Oh, M.H.; Cho, H.; Lee, J.H.; Lee, H.J.; Jang, S.H.; Lee, M.S. Loss of ARID1A Expression in Gastric Cancer: Correlation with Mismatch Repair Deficiency and Clinicopathologic Features. J. Gastric Cancer 2015, 15, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, S.; Kohashi, K.; Yamada, Y.; Fujiwara, M.; Koga, Y.; Ihara, E.; Ogawa, Y.; Oki, E.; Nakamura, M.; Oda, Y. Solid-type poorly differentiated adenocarcinoma of the stomach: Deficiency of mismatch repair and SWI/SNF complex. Cancer Sci. 2020, 111, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Xu, C.; Mandoli, A.; Okabe, A.; Chen, G.B.; Huang, K.K.; Sheng, T.; Yao, X.; Teo, J.M.N.; Sundar, R.; et al. Chromatin Rewiring by Mismatch Repair Protein MSH2 Alters Cell Adhesion Pathways and Sensitivity to BET Inhibition in Gastric Cancer. Cancer Res. 2022, 82, 2538–2551. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M.; Chastain, P.D., 2nd; Rosson, G.B.; Groh, B.S.; Weissman, B.E.; Kaufman, D.G.; Bultman, S.J. BRG1 co-localizes with DNA replication factors and is required for efficient replication fork progression. Nucleic Acids Res. 2010, 38, 6906–6919. [Google Scholar] [CrossRef] [PubMed]
- Bayona-Feliu, A.; Barroso, S.; Munoz, S.; Aguilera, A. The SWI/SNF chromatin remodeling complex helps resolve R-loop-mediated transcription-replication conflicts. Nat. Genet. 2021, 53, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.; Fournier, L.A.; Chang, E.Y.; Wells, J.P.; Minaker, S.W.; Zhu, Y.D.; Wang, A.Y.; Wang, Y.; Huntsman, D.G.; Stirling, P.C. ARID1A regulates R-loop associated DNA replication stress. PLoS Genet. 2021, 17, e1009238. [Google Scholar] [CrossRef]
- Gupta, M.; Concepcion, C.P.; Fahey, C.G.; Keshishian, H.; Bhutkar, A.; Brainson, C.F.; Sanchez-Rivera, F.J.; Pessina, P.; Kim, J.Y.; Simoneau, A.; et al. BRG1 Loss Predisposes Lung Cancers to Replicative Stress and ATR Dependency. Cancer Res. 2020, 80, 3841–3854. [Google Scholar] [CrossRef]
- Shain, A.H.; Pollack, J.R. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE 2013, 8, e55119. [Google Scholar] [CrossRef]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Tessema, M.; Weissman, B.E. The SWI/SNF Complex: A Frequently Mutated Chromatin Remodeling Complex in Cancer. In Epigenetics in Oncology; Chen, J., Wang, G.G., Lu, J., Eds.; Cancer Treatment and Research; Springer: Cham, Switzerland, 2023; Volume 190, pp. 211–244. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, J.; Tang, Y. Advances in the role of SWI/SNF complexes in tumours. J. Cell. Mol. Med. 2023, 27, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tang, J. SWI/SNF complexes and cancers. Gene 2023, 870, 147420. [Google Scholar] [CrossRef]
- Dreier, M.R.; de la Serna, I.L. SWI/SNF Chromatin Remodeling Enzymes in Melanoma. Epigenomes 2022, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, S.C.; Wei, Y.; Luo, X.; Jia, Y.; Li, L.; Gopal, P.; Zhu, M.; Nassour, I.; Chuang, J.C.; et al. Arid1a Has Context-Dependent Oncogenic and Tumor Suppressor Functions in Liver Cancer. Cancer Cell 2017, 32, 574–589.e6. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.W.; Hong, A.L. SMARCB1-Deficient Cancers: Novel Molecular Insights and Therapeutic Vulnerabilities. Cancers 2022, 14, 3645. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lee, R.S.; Alver, B.H.; Haswell, J.R.; Wang, S.; Mieczkowski, J.; Drier, Y.; Gillespie, S.M.; Archer, T.C.; Wu, J.N.; et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 2017, 49, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Kia, S.K.; Gorski, M.M.; Giannakopoulos, S.; Verrijzer, C.P. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell. Biol. 2008, 28, 3457–3464. [Google Scholar] [CrossRef]
- Walhart, T.A.; Vacca, B.; Hepperla, A.J.; Hamad, S.H.; Petrongelli, J.; Wang, Y.; McKean, E.L.; Moksa, M.; Cao, Q.; Yip, S.; et al. SMARCB1 Loss in Poorly Differentiated Chordomas Drives Tumor Progression. Am. J. Pathol. 2023, 193, 456–473. [Google Scholar] [CrossRef]
- Guidi, C.J.; Sands, A.T.; Zambrowicz, B.P.; Turner, T.K.; Demers, D.A.; Webster, W.; Smith, T.W.; Imbalzano, A.N.; Jones, S.N. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol. Cell. Biol. 2001, 21, 3598–3603. [Google Scholar] [CrossRef]
- Roberts, C.W.; Galusha, S.A.; McMenamin, M.E.; Fletcher, C.D.; Orkin, S.H. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 13796–13800. [Google Scholar] [CrossRef]
- Li, M.; Zhao, H.; Zhang, X.; Wood, L.D.; Anders, R.A.; Choti, M.A.; Pawlik, T.M.; Daniel, H.D.; Kannangai, R.; Offerhaus, G.J.; et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat. Genet. 2011, 43, 828–829. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Schoenfeld, D.A.; Zhou, R.; Zairis, S.; Su, W.; Steinbach, N.; Mathur, D.; Bansal, A.; Zachem, A.L.; Tavarez, B.; Hasson, D.; et al. Loss of PBRM1 Alters Promoter Histone Modifications and Activates ALDH1A1 to Drive Renal Cell Carcinoma. Mol. Cancer Res. 2022, 20, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Hong, J.H.; Nargund, A.M.; Ng, M.S.W.; Heng, H.L.; Li, Z.; Guan, P.; Sugiura, M.; Chu, P.L.; Wang, L.C.; et al. PBRM1-deficient PBAF complexes target aberrant genomic loci to activate the NF-kappaB pathway in clear cell renal cell carcinoma. Nat. Cell Biol. 2023, 25, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Alver, B.H.; Kim, K.H.; Lu, P.; Wang, X.; Manchester, H.E.; Wang, W.; Haswell, J.R.; Park, P.J.; Roberts, C.W. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 2017, 8, 14648. [Google Scholar] [CrossRef]
- Hodges, H.C.; Stanton, B.Z.; Cermakova, K.; Chang, C.Y.; Miller, E.L.; Kirkland, J.G.; Ku, W.L.; Veverka, V.; Zhao, K.; Crabtree, G.R. Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat. Struct. Mol. Biol. 2018, 25, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Parolia, A.; Qiao, Y.; Bawa, P.; Eyunni, S.; Mannan, R.; Carson, S.E.; Chang, Y.; Wang, X.; Zhang, Y.; et al. Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer. Nature 2022, 601, 434–439. [Google Scholar] [CrossRef]
- Wang, X.; Sansam, C.G.; Thom, C.S.; Metzger, D.; Evans, J.A.; Nguyen, P.T.; Roberts, C.W. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res. 2009, 69, 8094–8101. [Google Scholar] [CrossRef] [PubMed]
- McBride, M.J.; Pulice, J.L.; Beird, H.C.; Ingram, D.R.; D’Avino, A.R.; Shern, J.F.; Charville, G.W.; Hornick, J.L.; Nakayama, R.T.; Garcia-Rivera, E.M.; et al. The SS18-SSX Fusion Oncoprotein Hijacks BAF Complex Targeting and Function to Drive Synovial Sarcoma. Cancer Cell 2018, 33, 1128–1141.e7. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Liu, P.; Zhang, S.; Donahue, K.; Wang, Y.; Schehr, J.L.; Wolfe, S.K.; Dickerson, A.; Lu, L.; Rui, L.; et al. BAF155 methylation drives metastasis by hijacking super-enhancers and subverting anti-tumor immunity. Nucleic Acids Res. 2021, 49, 12211–12233. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, A.F.; Martin, L.J.; Minder, J.L.; Roe, J.-S.; Shi, J.; Steurer, S.; Bader, G.; McConnell, D.; Pearson, M.; Gerstberger, T.; et al. Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nat. Chem. Biol. 2016, 12, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.D.; Veenstra, H.; Khasnis, S.; Gunnell, A.; Webb, H.M.; Shannon-Lowe, C.; Andrews, S.; Osborne, C.S.; West, M.J. MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. eLife 2016, 5, e18270. [Google Scholar] [CrossRef] [PubMed]
- Keenen, B.; Qi, H.; Saladi, S.; Yeung, M.; De La Serna, I. Heterogeneous SWI/SNF chromatin remodeling complexes promote expression of microphthalmia-associated transcription factor target genes in melanoma. Oncogene 2010, 29, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, G.R.; Rahal, R.; Buxton, F.; Xiang, K.; McAllister, G.; Frias, E.; Bagdasarian, L.; Huber, J.; Lindeman, A.; Chen, D.; et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 3128–3133. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Helming, K.C.; Wang, X.; Kim, Y.; Vazquez, F.; Jagani, Z.; Hahn, W.C.; Roberts, C.W. Residual complexes containing SMARCA2 (BRM) underlie the oncogenic drive of SMARCA4 (BRG1) mutation. Mol. Cell. Biol. 2014, 34, 1136–1144. [Google Scholar] [CrossRef]
- Ding, Y.; Li, N.; Dong, B.; Guo, W.; Wei, H.; Chen, Q.; Yuan, H.; Han, Y.; Chang, H.; Kan, S.; et al. Chromatin remodeling ATPase BRG1 and PTEN are synthetic lethal in prostate cancer. J. Clin. Investig. 2019, 129, 759–773. [Google Scholar] [CrossRef]
- Xue, Y.; Meehan, B.; Fu, Z.; Wang, X.Q.D.; Fiset, P.O.; Rieker, R.; Levins, C.; Kong, T.; Zhu, X.; Morin, G.; et al. SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nat. Commun. 2019, 10, 557. [Google Scholar] [CrossRef]
- Fukumoto, T.; Park, P.H.; Wu, S.; Fatkhutdinov, N.; Karakashev, S.; Nacarelli, T.; Kossenkov, A.V.; Speicher, D.W.; Jean, S.; Zhang, L.; et al. Repurposing Pan-HDAC Inhibitors for ARID1A-Mutated Ovarian Cancer. Cell Rep. 2018, 22, 3393–3400. [Google Scholar] [CrossRef]
- Cheng, X.; Zhao, J.X.; Dong, F.; Cao, X.C. ARID1A Mutation in Metastatic Breast Cancer: A Potential Therapeutic Target. Front. Oncol. 2021, 11, 759577. [Google Scholar] [CrossRef]
- Kuo, T.L.; Cheng, K.H.; Chen, L.T.; Hung, W.C. ARID1A loss in pancreas leads to islet developmental defect and metabolic disturbance. iScience 2023, 26, 105881. [Google Scholar] [CrossRef]
- Romero, O.A.; Vilarrubi, A.; Alburquerque-Bejar, J.J.; Gomez, A.; Andrades, A.; Trastulli, D.; Pros, E.; Setien, F.; Verdura, S.; Farre, L.; et al. SMARCA4 deficient tumours are vulnerable to KDM6A/UTX and KDM6B/JMJD3 blockade. Nat. Commun. 2021, 12, 4319. [Google Scholar] [CrossRef]
- Xue, Y.; Meehan, B.; Macdonald, E.; Venneti, S.; Wang, X.Q.D.; Witkowski, L.; Jelinic, P.; Kong, T.; Martinez, D.; Morin, G.; et al. CDK4/6 inhibitors target SMARCA4-determined cyclin D1 deficiency in hypercalcemic small cell carcinoma of the ovary. Nat. Commun. 2019, 10, 558. [Google Scholar] [CrossRef]
- Dillon, M.T.; Guevara, J.; Mohammed, K.; Patin, E.C.; Smith, S.A.; Dean, E.; Jones, G.N.; Willis, S.E.; Petrone, M.; Silva, C.; et al. Durable responses to ATR inhibition with ceralasertib in tumors with genomic defects and high inflammation. J. Clin. Investig. 2023, 134, e175369. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C.; Antonelli, R.; Nadal-Ribelles, M.; Devis-Jauregui, L.; Latorre, P.; Sole, C.; Masanas, M.; Molero-Valenzuela, A.; Soriano, A.; Sanchez de Toledo, J.; et al. Structural disruption of BAF chromatin remodeller impairs neuroblastoma metastasis by reverting an invasiveness epigenomic program. Mol. Cancer 2022, 21, 175. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, J.; Wu, J.; Xu, B.; Wang, Z.; Giamas, G.; Stebbing, J.; Yu, Z. A Pan-Cancer Analysis of SMARCA4 Alterations in Human Cancers. Front. Immunol. 2021, 12, 762598. [Google Scholar] [CrossRef] [PubMed]
- Mason, L.D.; Chava, S.; Reddi, K.K.; Gupta, R. The BRD9/7 Inhibitor TP-472 Blocks Melanoma Tumor Growth by Suppressing ECM-Mediated Oncogenic Signaling and Inducing Apoptosis. Cancers 2021, 13, 5516. [Google Scholar] [CrossRef]
- Chory, E.J.; Kirkland, J.G.; Chang, C.Y.; D’Andrea, V.D.; Gourisankar, S.; Dykhuizen, E.C.; Crabtree, G.R. Chemical Inhibitors of a Selective SWI/SNF Function Synergize with ATR Inhibition in Cancer Cell Killing. ACS Chem. Biol. 2020, 15, 1685–1696. [Google Scholar] [CrossRef]
- Guo, A.; Huang, H.; Zhu, Z.; Chen, M.J.; Shi, H.; Yuan, S.; Sharma, P.; Connelly, J.P.; Liedmann, S.; Dhungana, Y.; et al. cBAF complex components and MYC cooperate early in CD8(+) T cell fate. Nature 2022, 607, 135–141. [Google Scholar] [CrossRef]
- Chamberlain, P.P.; Hamann, L.G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15, 937–944. [Google Scholar] [CrossRef]
- Collins, M.; Thomsen, A.; Gartin, A.; Sandoval, G.J.; Adam, A.; Reilly, S.; Delestre, L.; Penard-Lacronique, V.; Fiskus, W.; Bhalla, K.; et al. Abstract 2122: The dual BRM/BRG1 (SMARCA2/4) inhibitor FHD-286 induces differentiation in preclinical models of AML. Proceedings of the American Association for Cancer Research Annual Meeting 2023. Cancer Res. 2023, 83 (Suppl. S7), 2122. [Google Scholar] [CrossRef]
- Hentemann, M. Abstract ND14: Pharmacological profile and anti-tumor properties of FHD-286: A novel BAF inhibitor for the treatment of transcription factor-driven cancers. Proceedings of the American Association for Cancer Research Annual Meeting 2022. Cancer Res. 2022, 82 (Suppl. S12), ND14. [Google Scholar] [CrossRef]
- Hulse, M.; Agarwal, A.; Wang, M.; Carter, J.; Sivakumar, M.; Vidal, B.; Brown, J.; Moore, A.; Grego, A.; Bhagwat, N.; et al. Abstract 3263: Preclinical characterization of PRT3789, a potent and selective SMARCA2 targeted degrader. Proceedings of the American Association for Cancer Research Annual Meeting 2022. Cancer Res. 2022, 82, 3263. [Google Scholar] [CrossRef]
- Dominici, C.; Mayhew, D.; Adam, A.; Uzan, F.; Garbitt-Amaral, V.; Mikse, O.; Antonakos, B.; Ahmad, H.; Parikh, S.; Lin, M.Y.; et al. Abstract A049: Investigation of FHD-609, a potent degrader of BRD9, in preclinical models of acute myeloid leukemia (AML). Proceedings of the AACR-NCI-EORTC Virtual International Conference on Molecular Targets and Cancer Therapeutics. Mol. Cancer Ther. 2023, 22 (Suppl. S12), A049. [Google Scholar] [CrossRef]
- Jackson, K.L.; Agafonov, R.V.; Carlson, M.W.; Chaturvedi, P.; Cocozziello, D.; Cole, K.; Deibler, R.; Eron, S.J.; Good, A.; Hart, A.A.; et al. Abstract ND09: The discovery and characterization of CFT8634: A potent and selective degrader of BRD9 for the treatment of SMARCB1-perturbed cancers. Proceedings of the American Association for Cancer Research Annual Meeting 2022. Cancer Res. 2022, 82, ND09. [Google Scholar] [CrossRef]
- Dutta, P.; Tanti, G.K.; Sharma, S.; Goswami, S.K.; Komath, S.S.; Mayo, M.W.; Hockensmith, J.W.; Muthuswami, R. Global epigenetic changes induced by SWI2/SNF2 inhibitors characterize neomycin-resistant mammalian cells. PLoS ONE 2012, 7, e49822. [Google Scholar] [CrossRef] [PubMed]
- Muthuswami, R.; Mesner, L.D.; Wang, D.; Hill, D.A.; Imbalzano, A.N.; Hockensmith, J.W. Phosphoaminoglycosides inhibit SWI2/SNF2 family DNA-dependent molecular motor domains. Biochemistry 2000, 39, 4358–4365. [Google Scholar] [CrossRef] [PubMed]
- Felle, M.; Exler, J.H.; Merkl, R.; Dachauer, K.; Brehm, A.; Grummt, I.; Langst, G. DNA sequence encoded repression of rRNA gene transcription in chromatin. Nucleic Acids Res. 2010, 38, 5304–5314. [Google Scholar] [CrossRef]
- Muthuswami, R.; Bailey, L.; Rakesh, R.; Imbalzano, A.N.; Nickerson, J.A.; Hockensmith, J.W. BRG1 is a prognostic indicator and a potential therapeutic target for prostate cancer. J. Cell. Physiol. 2019, 234, 15194–15205. [Google Scholar] [CrossRef]
- Rakesh, R.; Chanana, U.B.; Hussain, S.; Sharma, S.; Goel, K.; Bisht, D.; Patne, K.; Swer, P.B.; Hockensmith, J.W.; Muthuswami, R. Altering mammalian transcription networking with ADAADi: An inhibitor of ATP-dependent chromatin remodeling. PLoS ONE 2021, 16, e0251354. [Google Scholar] [CrossRef]
- Papillon, J.P.N.; Nakajima, K.; Adair, C.D.; Hempel, J.; Jouk, A.O.; Karki, R.G.; Mathieu, S.; Mobitz, H.; Ntaganda, R.; Smith, T.; et al. Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)/SMARCA4-Mutant Cancers. J. Med. Chem. 2018, 61, 10155–10172. [Google Scholar] [CrossRef]
- Panditharatna, E.; Marques, J.G.; Wang, T.; Trissal, M.C.; Liu, I.; Jiang, L.; Beck, A.; Groves, A.; Dharia, N.V.; Li, D.; et al. BAF Complex Maintains Glioma Stem Cells in Pediatric H3K27M Glioma. Cancer Discov. 2022, 12, 2880–2905. [Google Scholar] [CrossRef]
- Mo, Y.; Duan, S.; Zhang, X.; Hua, X.; Zhou, H.; Wei, H.J.; Watanabe, J.; McQuillan, N.; Su, Z.; Gu, W.; et al. Epigenome Programming by H3.3K27M Mutation Creates a Dependence of Pediatric Glioma on SMARCA4. Cancer Discov. 2022, 12, 2906–2929. [Google Scholar] [CrossRef]
- Rago, F.; Rodrigues, L.U.; Bonney, M.; Sprouffske, K.; Kurth, E.; Elliott, G.; Ambrose, J.; Aspesi, P.; Oborski, J.; Chen, J.T.; et al. Exquisite Sensitivity to Dual BRG1/BRM ATPase Inhibitors Reveals Broad SWI/SNF Dependencies in Acute Myeloid Leukemia. Mol. Cancer Res. 2022, 20, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Rago, F.; Elliott, G.; Li, A.; Sprouffske, K.; Kerr, G.; Desplat, A.; Abramowski, D.; Chen, J.T.; Farsidjani, A.; Xiang, K.X.; et al. The Discovery of SWI/SNF Chromatin Remodeling Activity as a Novel and Targetable Dependency in Uveal Melanoma. Mol. Cancer Ther. 2020, 19, 2186–2195. [Google Scholar] [CrossRef] [PubMed]
- Smit, K.N.; Jager, M.J.; de Klein, A.; Kiliҫ, E. Uveal melanoma: Towards a molecular understanding. Prog. Retin. Eye Res. 2020, 75, 100800. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Horn, P.J.; Peterson, C.L. The bromodomain: A regulator of ATP-dependent chromatin remodeling? Front. Biosci. 2001, 6, 1019–1023. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Fedorov, O.; Keller, M.; Wrobel, M.; Morgenstern, O.; Bracher, F.; Knapp, S. Benzodiazepines and benzotriazepines as protein interaction inhibitors targeting bromodomains of the BET family. Bioorganic Med. Chem. 2012, 20, 1878–1886. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Fish, P.V.; Filippakopoulos, P.; Bish, G.; Brennan, P.E.; Bunnage, M.E.; Cook, A.S.; Federov, O.; Gerstenberger, B.S.; Jones, H.; Knapp, S.; et al. Identification of a chemical probe for bromo and extra C-terminal bromodomain inhibition through optimization of a fragment-derived hit. J. Med. Chem. 2012, 55, 9831–9837. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Filippakopoulos, P.; Knapp, S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, e29. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Da Costa, D.; Thanasopoulou, A.; Filippakopoulos, P.; Fish, P.V.; Philpott, M.; Fedorov, O.; Brennan, P.; Bunnage, M.E.; Owen, D.R.; et al. PFI-1, a highly selective protein interaction inhibitor, targeting BET Bromodomains. Cancer Res. 2013, 73, 3336–3346. [Google Scholar] [CrossRef] [PubMed]
- Clegg, M.A.; Tomkinson, N.C.O.; Prinjha, R.K.; Humphreys, P.G. Advancements in the Development of non-BET Bromodomain Chemical Probes. ChemMedChem 2019, 14, 362–385. [Google Scholar] [CrossRef]
- Singh, M.; Popowicz, G.M.; Krajewski, M.; Holak, T.A. Structural ramification for acetyl-lysine recognition by the bromodomain of human BRG1 protein, a central ATPase of the SWI/SNF remodeling complex. ChemBioChem 2007, 8, 1308–1316. [Google Scholar] [CrossRef]
- Lloyd, J.T.; Glass, K.C. Biological function and histone recognition of family IV bromodomain-containing proteins. J. Cell. Physiol. 2018, 233, 1877–1886. [Google Scholar] [CrossRef]
- Gerstenberger, B.S.; Trzupek, J.D.; Tallant, C.; Fedorov, O.; Filippakopoulos, P.; Brennan, P.E.; Fedele, V.; Martin, S.; Picaud, S.; Rogers, C.; et al. Identification of a Chemical Probe for Family VIII Bromodomains through Optimization of a Fragment Hit. J. Med. Chem. 2016, 59, 4800–4811. [Google Scholar] [CrossRef]
- Fedorov, O.; Castex, J.; Tallant, C.; Owen, D.R.; Martin, S.; Aldeghi, M.; Monteiro, O.; Filippakopoulos, P.; Picaud, S.; Trzupek, J.D.; et al. Selective targeting of the BRG/PB1 bromodomains impairs embryonic and trophoblast stem cell maintenance. Sci. Adv. 2015, 1, e1500723. [Google Scholar] [CrossRef]
- Sharma, T.; Olea-Flores, M.; Imbalzano, A.N. Regulation of the Wnt signaling pathway during myogenesis by the mammalian SWI/SNF ATPase BRG1. Front. Cell Dev. Biol. 2023, 11, 1160227. [Google Scholar] [CrossRef]
- Sharma, T.; Robinson, D.C.L.; Witwicka, H.; Dilworth, F.J.; Imbalzano, A.N. The Bromodomains of the mammalian SWI/SNF (mSWI/SNF) ATPases Brahma (BRM) and Brahma Related Gene 1 (BRG1) promote chromatin interaction and are critical for skeletal muscle differentiation. Nucleic Acids Res. 2021, 49, 8060–8077. [Google Scholar] [CrossRef]
- Basuroy, T.; Dreier, M.; Baum, C.; Blomquist, T.; Trumbly, R.; Filipp, F.V.; de la Serna, I.L. Epigenetic and pharmacological control of pigmentation via Bromodomain Protein 9 (BRD9). Pigment. Cell Melanoma Res. 2022, 36, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Sharma, S.; Cui, H.; LeBlanc, S.E.; Zhang, H.; Muthuswami, R.; Nickerson, J.A.; Imbalzano, A.N. Targeting the chromatin remodeling enzyme BRG1 increases the efficacy of chemotherapy drugs in breast cancer cells. Oncotarget 2016, 7, 27158–27175. [Google Scholar] [CrossRef] [PubMed]
- Vangamudi, B.; Paul, T.A.; Shah, P.K.; Kost-Alimova, M.; Nottebaum, L.; Shi, X.; Zhan, Y.; Leo, E.; Mahadeshwar, H.S.; Protopopov, A.; et al. The SMARCA2/4 ATPase Domain Surpasses the Bromodomain as a Drug Target in SWI/SNF-Mutant Cancers: Insights from cDNA Rescue and PFI-3 Inhibitor Studies. Cancer Res. 2015, 75, 3865–3878. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.S.Y.; Chooi, J.Y.; Lim, J.S.L.; Toh, S.H.M.; Tan, T.Z.; Chng, W.J. SMARCA2 Is a Novel Interactor of NSD2 and Regulates Prometastatic PTP4A3 through Chromatin Remodeling in t(4;14) Multiple Myeloma. Cancer Res. 2021, 81, 2332–2344. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, Y.; Sims, M.M.; He, Y.; Miller, D.D.; Pfeffer, L.M. Targeting the Bromodomain of BRG-1/BRM Subunit of the SWI/SNF Complex Increases the Anticancer Activity of Temozolomide in Glioblastoma. Pharmaceuticals 2021, 14, 904. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Lee, D.Y.; Hwang, Y.S.; Seo, H.R.; Lee, S.A.; Kwon, J. The Bromodomain Inhibitor PFI-3 Sensitizes Cancer Cells to DNA Damage by Targeting SWI/SNF. Mol. Cancer Res. 2021, 19, 900–912. [Google Scholar] [CrossRef]
- Yang, C.; He, Y.; Wang, Y.; McKinnon, P.J.; Shahani, V.; Miller, D.D.; Pfeffer, L.M. Next-generation bromodomain inhibitors of the SWI/SNF complex enhance DNA damage and cell death in glioblastoma. J. Cell. Mol. Med. 2023, 27, 2770–2781. [Google Scholar] [CrossRef]
- Wanior, M.; Preuss, F.; Ni, X.; Kramer, A.; Mathea, S.; Gobel, T.; Heidenreich, D.; Simonyi, S.; Kahnt, A.S.; Joerger, A.C.; et al. Pan-SMARCA/PB1 Bromodomain Inhibitors and Their Role in Regulating Adipogenesis. J. Med. Chem. 2020, 63, 14680–14699. [Google Scholar] [CrossRef]
- Melin, L.; Gesner, E.; Attwell, S.; Kharenko, O.A.; van der Horst, E.H.; Hansen, H.C.; Gagnon, A. Design and Synthesis of LM146, a Potent Inhibitor of PB1 with an Improved Selectivity Profile over SMARCA2. ACS Omega 2021, 6, 21327–21338. [Google Scholar] [CrossRef]
- Shishodia, S.; Nunez, R.; Strohmier, B.P.; Bursch, K.L.; Goetz, C.J.; Olp, M.D.; Jensen, D.R.; Fenske, T.G.; Ordonez-Rubiano, S.C.; Blau, M.E.; et al. Selective and Cell-Active PBRM1 Bromodomain Inhibitors Discovered through NMR Fragment Screening. J. Med. Chem. 2022, 65, 13714–13735. [Google Scholar] [CrossRef]
- Cochran, A.G.; Flynn, M. GNE-235: A Lead Compound Selective for the Second Bromodomain of PBRM1. J. Med. Chem. 2023, 66, 13116–13134. [Google Scholar] [CrossRef]
- Hopson, S.; Thompson, M.J. BAF180: Its Roles in DNA Repair and Consequences in Cancer. ACS Chem. Biol. 2017, 12, 2482–2490. [Google Scholar] [CrossRef] [PubMed]
- Mota, S.T.S.; Vecchi, L.; Zoia, M.A.P.; Oliveira, F.M.; Alves, D.A.; Dornelas, B.C.; Bezerra, S.M.; Andrade, V.P.; Maia, Y.C.P.; Neves, A.F.; et al. New Insights into the Role of Polybromo-1 in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 2852. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, M.; Fushimi, A.; Yamashita, N.; Bhattacharya, A.; Rajabi, H.; Long, M.D.; Yasumizu, Y.; Oya, M.; Liu, S.; Kufe, D. MUC1-C activates the PBAF chromatin remodeling complex in integrating redox balance with progression of human prostate cancer stem cells. Oncogene 2021, 40, 4930–4940. [Google Scholar] [CrossRef] [PubMed]
- Sutherell, C.L.; Tallant, C.; Monteiro, O.P.; Yapp, C.; Fuchs, J.E.; Fedorov, O.; Siejka, P.; Muller, S.; Knapp, S.; Brenton, J.D.; et al. Identification and Development of 2,3-Dihydropyrrolo [1,2-a]quinazolin-5(1H)-one Inhibitors Targeting Bromodomains within the Switch/Sucrose Nonfermenting Complex. J. Med. Chem. 2016, 59, 5095–5101. [Google Scholar] [CrossRef] [PubMed]
- Bharathy, N.; Cleary, M.M.; Kim, J.A.; Nagamori, K.; Crawford, K.A.; Wang, E.; Saha, D.; Settelmeyer, T.P.; Purohit, R.; Skopelitis, D.; et al. SMARCA4 biology in alveolar rhabdomyosarcoma. Oncogene 2022, 41, 1647–1656. [Google Scholar] [CrossRef]
- Mota, M.; Sweha, S.R.; Pun, M.; Natarajan, S.K.; Ding, Y.; Chung, C.; Hawes, D.; Yang, F.; Judkins, A.R.; Samajdar, S.; et al. Targeting SWI/SNF ATPases in H3.3K27M diffuse intrinsic pontine gliomas. Proc. Natl. Acad. Sci. USA 2023, 120, e2221175120. [Google Scholar] [CrossRef]
- Cantley, J.; Ye, X.; Rousseau, E.; Januario, T.; Hamman, B.D.; Rose, C.M.; Cheung, T.K.; Hinkle, T.; Soto, L.; Quinn, C.; et al. Selective PROTAC-mediated degradation of SMARCA2 is efficacious in SMARCA4 mutant cancers. Nat. Commun. 2022, 13, 6814. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25, 78–87.e5. [Google Scholar] [CrossRef]
- Li, M.; Wei, Y.; Liu, Y.; Wei, J.; Zhou, X.; Duan, Y.; Chen, S.; Xue, C.; Zhan, Y.; Zheng, L.; et al. BRD7 inhibits enhancer activity and expression of BIRC2 to suppress tumor growth and metastasis in nasopharyngeal carcinoma. Cell Death Dis. 2023, 14, 121. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, R.; Wang, H.; Luo, Y.; Wang, X.; Niu, W.; Zhou, Y.; Wen, Q.; Fan, S.; Li, X.; et al. miR-141 is involved in BRD7-mediated cell proliferation and tumor formation through suppression of the PTEN/AKT pathway in nasopharyngeal carcinoma. Cell Death Dis. 2016, 7, e2156. [Google Scholar] [CrossRef]
- Jin, J.; Chen, F.; He, W.; Zhao, L.; Lin, B.; Zheng, D.; Chen, L.; He, H.; He, Q. YAP-Activated SATB2 Is a Coactivator of NRF2 That Amplifies Antioxidative Capacity and Promotes Tumor Progression in Renal Cell Carcinoma. Cancer Res. 2023, 83, 786–803. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Liu, D.; Zhao, Y. BRD7 suppresses tumor chemosensitivity to CHK1 inhibitors by inhibiting USP1-mediated deubiquitination of CHK1. Cell Death Discov. 2023, 9, 313. [Google Scholar] [CrossRef]
- Clark, P.G.; Vieira, L.C.; Tallant, C.; Fedorov, O.; Singleton, D.C.; Rogers, C.M.; Monteiro, O.P.; Bennett, J.M.; Baronio, R.; Muller, S.; et al. LP99: Discovery and Synthesis of the First Selective BRD7/9 Bromodomain Inhibitor. Angew. Chem. 2015, 54, 6217–6221. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.R.; Burmeister, A.; Skowron, M.A.; Stephan, A.; Bremmer, F.; Wakileh, G.A.; Petzsch, P.; Kohrer, K.; Albers, P.; Nettersheim, D. Therapeutical interference with the epigenetic landscape of germ cell tumors: A comparative drug study and new mechanistical insights. Clin. Epigenetics 2022, 14, 5. [Google Scholar] [CrossRef]
- Hugle, M.; Regenass, P.; Warstat, R.; Hau, M.; Schmidtkunz, K.; Lucas, X.; Wohlwend, D.; Einsle, O.; Jung, M.; Breit, B.; et al. 4-Acyl Pyrroles as Dual BET-BRD7/9 Bromodomain Inhibitors Address BETi Insensitive Human Cancer Cell Lines. J. Med. Chem. 2020, 63, 15603–15620. [Google Scholar] [CrossRef]
- Martin, L.J.; Koegl, M.; Bader, G.; Cockcroft, X.L.; Fedorov, O.; Fiegen, D.; Gerstberger, T.; Hofmann, M.H.; Hohmann, A.F.; Kessler, D.; et al. Structure-Based Design of an in Vivo Active Selective BRD9 Inhibitor. J. Med. Chem. 2016, 59, 4462–4475. [Google Scholar] [CrossRef]
- Theodoulou, N.H.; Bamborough, P.; Bannister, A.J.; Becher, I.; Bit, R.A.; Che, K.H.; Chung, C.W.; Dittmann, A.; Drewes, G.; Drewry, D.H.; et al. Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J. Med. Chem. 2016, 59, 1425–1439. [Google Scholar] [CrossRef]
- Zhou, L.; Yao, Q.; Li, H.; Chen, J. Targeting BRD9 by I-BRD9 efficiently inhibits growth of acute myeloid leukemia cells. Transl Cancer Res. 2021, 10, 3364–3372. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, L.; Lou, W.; Su, J.; Huang, J.; Liu, A.; Xu, Y.; He, H.; Gao, Y.; Xu, D.; et al. Aberrant activation of m6A demethylase FTO renders HIF2alpha(low/-) clear cell renal cell carcinoma sensitive to BRD9 inhibitors. Sci. Transl. Med. 2021, 13, eabf6045. [Google Scholar] [CrossRef]
- Zhu, Q.; Gu, X.; Wei, W.; Wu, Z.; Gong, F.; Dong, X. BRD9 is an essential regulator of glycolysis that creates an epigenetic vulnerability in colon adenocarcinoma. Cancer Med. 2023, 12, 1572–1587. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, S.; Damiani, E.; Wang, S.; Dharmanand, R.; Tripathi, C.; Tovar Perez, J.E.; Dashwood, W.M.; Rajendran, P.; Dashwood, R.H. BRD9 Inhibition by Natural Polyphenols Targets DNA Damage/Repair and Apoptosis in Human Colon Cancer Cells. Nutrients 2022, 14, 4317. [Google Scholar] [CrossRef]
- Yang, Q.; Bariani, M.V.; Falahati, A.; Khosh, A.; Lastra, R.R.; Siblini, H.; Boyer, T.G.; Al-Hendy, A. The Functional Role and Regulatory Mechanism of Bromodomain-Containing Protein 9 in Human Uterine Leiomyosarcoma. Cells 2022, 11, 2160. [Google Scholar] [CrossRef] [PubMed]
- Barghout, S.H.; Mann, M.K.; Aman, A.; Yu, Y.; Alteen, M.G.; Schimmer, A.D.; Schapira, M.; Arrowsmith, C.H.; Barsyte-Lovejoy, D. Combinatorial Anticancer Drug Screen Identifies Off-Target Effects of Epigenetic Chemical Probes. ACS Chem. Biol. 2022, 17, 2801–2816. [Google Scholar] [CrossRef] [PubMed]
- Mancarella, C.; Morrione, A.; Scotlandi, K. PROTAC-Based Protein Degradation as a Promising Strategy for Targeted Therapy in Sarcomas. Int. J. Mol. Sci. 2023, 24, 16346. [Google Scholar] [CrossRef]
- Remillard, D.; Buckley, D.L.; Paulk, J.; Brien, G.L.; Sonnett, M.; Seo, H.S.; Dastjerdi, S.; Wuhr, M.; Dhe-Paganon, S.; Armstrong, S.A.; et al. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew. Chem. 2017, 56, 5738–5743. [Google Scholar] [CrossRef]
- Brien, G.L.; Remillard, D.; Shi, J.; Hemming, M.L.; Chabon, J.; Wynne, K.; Dillon, E.T.; Cagney, G.; Van Mierlo, G.; Baltissen, M.P.; et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. eLife 2018, 7, e41305. [Google Scholar] [CrossRef]
- Kurata, K.; Samur, M.K.; Liow, P.; Wen, K.; Yamamoto, L.; Liu, J.; Morelli, E.; Gulla, A.; Tai, Y.T.; Qi, J.; et al. BRD9 Degradation Disrupts Ribosome Biogenesis in Multiple Myeloma. Clin. Cancer Res. 2023, 29, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Duan, H.; Gui, R.; Wu, M.; Shen, L.; Jin, Y.; Pang, A.; Yu, X.; Zeng, S.; Zhang, B.; et al. Structure-based identification of new orally bioavailable BRD9-PROTACs for treating acute myelocytic leukemia. Eur. J. Med. Chem. 2023, 262, 115872. [Google Scholar] [CrossRef]
- Zoppi, V.; Hughes, S.J.; Maniaci, C.; Testa, A.; Gmaschitz, T.; Wieshofer, C.; Koegl, M.; Riching, K.M.; Daniels, D.L.; Spallarossa, A.; et al. Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel-Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem. 2019, 62, 699–726. [Google Scholar] [CrossRef] [PubMed]
- Ordonez-Rubiano, S.C.; Maschinot, C.A.; Wang, S.; Sood, S.; Baracaldo-Lancheros, L.F.; Strohmier, B.P.; McQuade, A.J.; Smith, B.C.; Dykhuizen, E.C. Rational Design and Development of Selective BRD7 Bromodomain Inhibitors and Their Activity in Prostate Cancer. J. Med. Chem. 2023, 66, 11250–11270. [Google Scholar] [CrossRef]
- Pan, J.; McKenzie, Z.M.; D’Avino, A.R.; Mashtalir, N.; Lareau, C.A.; St Pierre, R.; Wang, L.; Shilatifard, A.; Kadoch, C. The ATPase module of mammalian SWI/SNF family complexes mediates subcomplex identity and catalytic activity-independent genomic targeting. Nat. Genet. 2019, 51, 618–626. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug Name | Mechanism of Action | Cancer Models in Which Drug Is Effective |
|---|---|---|
| ADAADi | Inhibitor of DNA-dependent ATPAse activity | Prostate, breast, hepatoblastoma, lung cancers, and HeLa cells |
| BRM014 | Allosteric inhibitor of SMARCA4/2 catalytic activity | Glioma, leukemia |
| BRM011 | Allosteric inhibitor of SMARCA4/2 catalytic activity | Leukemia |
| JQ-dS-4 | PROTAC: CRBN-based allosteric inhibitor of SMARCA4/2 catalytic activity | Glioma |
| FHD-286 | Allosteric inhibitor of SMARCA4/2 catalytic activity | Leukemia, uveal melanoma |
| PFI-3 | Class VIII-selective SWI/SNF bromodomain inhibitor: SMARCA4/2, PBRM1 | Multiple myeloma, glioblastoma when combined with temozolomide |
| PB16 | PBRM1-selective bromodomain inhibitor | Prostate cancer |
| GNE-235 | PBRM1-selective bromodomain inhibitor | Not yet tested |
| ACBI1 | PROTAC: VHL-based Class VIII bromodomain-selective ligand that degrades SMARCA4/2 and PBRM1 | Alveolar rhabdomyosarcoma, leukemia |
| AU15330 | PROTAC: VHL-based Class VIII bromodomain-selective ligand that degrades SMARCA4/2 and PBRM1 | Prostate, glioma, breast cancers, multiple myeloma, lymphoma, EWING sarcoma, SMARCA4-null melanoma, synergistic with enzalutamide in prostate cancer |
| A947 | PROTAC: VHL-based Class VIII bromodomain-selective ligand that selectively degrades SMARCA2 | Lung cancer |
| LP99 | Class IV bromodomain inhibitor that inhibits BRD9 and BRD7 | Germ tumor cells |
| BI-7271 | Class IV bromodomain inhibitor that selectively inhibits BRD9 | Leukemia |
| BI-7273 | Class IV bromodomain inhibitor that selectively inhibits BRD9 | Leukemia |
| BI-9564 | Class IV bromodomain inhibitor that selectively inhibits BRD9 | Leukemia |
| I-BRD9 | Class IV bromodomain inhibitor that selectively inhibits BRD9 | Leukemia, clear cell renal carcinoma, colorectal cancer, ovarian cancer in combination with DNA damaging agents |
| T-472 | Class IV bromodomain inhibitor that selectively inhibits BRD9 | Melanoma and uterine leiomyosarcoma |
| dBRD9 | PROTAC: CRBN-based Class VIII bromodomain-selective ligand (uses BI7273) that degrades BRD9 | Leukemia, synovial sarcoma, multiple myeloma, clear cell meningioma |
| C6 | PROTAC: CRBN-based Class VIII bromodomain-selective ligand (uses BI7271) that degrades BRD9 | Leukemia |
| VZ-185 | PROTAC: VHL-based Class VIII bromodomain-selective ligand that degrades BRD7 and BRD9 | Leukemia, malignant rhabdoid tumor |
| CFT8634 | PROTAC: CRBN-based Class VIII bromodomain -elective ligand that degrades BRD9 | Synovial sarcoma and soft-tissue sarcomas null for SMARCB1 |
| FHD-609 | PROTAC: CRBN-based Class VIII bromodomain-selective ligand that degrades BRD9 | Synovial sarcoma and soft-tissue sarcomas null for SMARCB1 |
| I-78 | Class IV bromodomain inhibitor that selectively inhibits BRD7 | Prostate cancer |
| 2-77 | Class IV bromodomain inhibitor that selectively inhibits BRD7 | Prostate cancer |
| BD98 | cBAF inhibitor, binds to cBAF complexes, and de-represses BMI1 | Colon and breast cancer cells when combined with ATR inhibitor |
| Study Title | NCT Number | Phase/Status | Conditions | Drug/Intervention: | Mechanism | References |
|---|---|---|---|---|---|---|
| FHD-286 as Monotherapy or Combination Therapy in Subjects With Advanced Hematologic Malignancies | NCT04891757 | Phase 1/recruiting | Advanced hematologic malignancies like R/R AML, R/R MDS, and R/R CMML not in blast crisis | FHD-286, low-dose cytarabine, decitabine | Selective, oral inhibitor of SMARCA4/2 | [125] |
| FHD-286 in Subjects With Metastatic Uveal Melanoma | NCT04879017 | Phase 1/active, not recruiting | Metastatic uveal melanoma | FHD-286 | Selective, oral inhibitor of SMARCA4/2 | [126] |
| A Study of PRT3789 in Participants With Select Advanced or Metastatic Solid Tumors With a SMARCA4 Mutation | NCT05639751 | Phase 1/recruiting | Advanced, recurrent, or metastatic solid tumor malignancies with loss of SMARCA4 due to truncating mutation and/or deletion without concomitant SMARCA2 mutation or loss of SMARCA2 protein expression | PRT3789 | Smarca2-bromodomain- binding degrader | [127] |
| FHD-609 in Subjects With Advanced Synovial Sarcoma or Advanced SMARCB1-Loss Tumors | NCT04965753 | Phase 1/active, not recruiting | Advanced synovial sarcoma or advanced SMARCB1-loss tumors | FHD-609 | Intravenously administered agent that binds the BRD9 bromodomain and leads to degradation of the BRD9 protein | [128] |
| A Study to Assess the Safety and Tolerability of CFT8634 in Locally Advanced or Metastatic SMARCB1-Perturbed Cancers, Including Synovial Sarcoma and SMARCB1-Null Tumors | NCT05355753 | Phase 1, Phase 2/active, not recruiting | Synovial or soft tissue sarcoma and SMARCB1-null tumors who have received prior systemic therapy, have relapsed/refractory tumors, have unresectable or metastatic disease, and are not candidates for available therapies known to confer clinical benefit | CFT8634 | Orally taken agent that binds the BRD9 bromodomain and leads to BRD9 degradation | [129] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dreier, M.R.; Walia, J.; de la Serna, I.L. Targeting SWI/SNF Complexes in Cancer: Pharmacological Approaches and Implications. Epigenomes 2024, 8, 7. https://doi.org/10.3390/epigenomes8010007
Dreier MR, Walia J, de la Serna IL. Targeting SWI/SNF Complexes in Cancer: Pharmacological Approaches and Implications. Epigenomes. 2024; 8(1):7. https://doi.org/10.3390/epigenomes8010007
Chicago/Turabian StyleDreier, Megan R., Jasmine Walia, and Ivana L. de la Serna. 2024. "Targeting SWI/SNF Complexes in Cancer: Pharmacological Approaches and Implications" Epigenomes 8, no. 1: 7. https://doi.org/10.3390/epigenomes8010007