
Population Epigenetics: The Extent of DNA Methylation Variation in Wild Animal Populations


Abstract
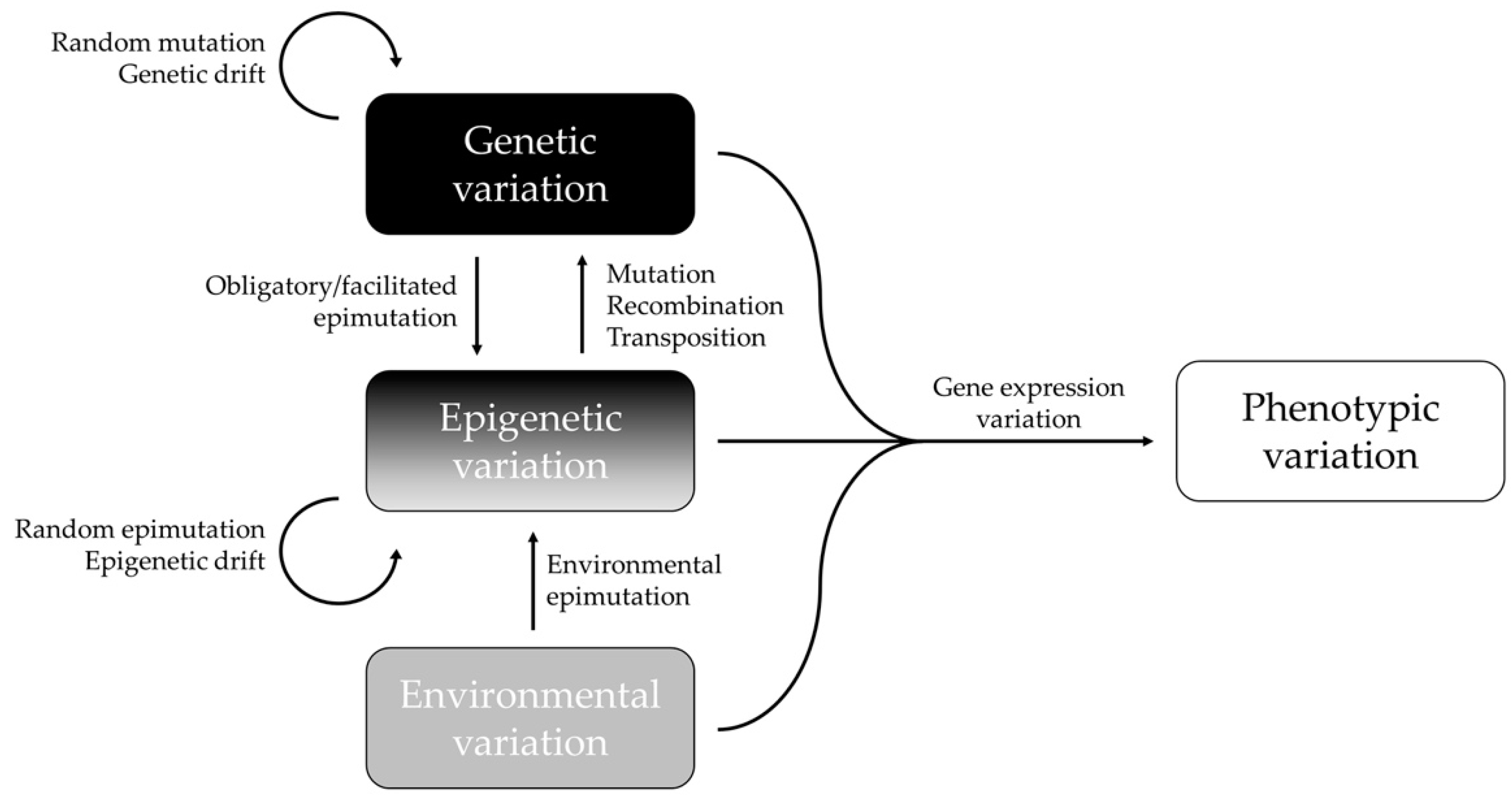
:1. Introduction
2. Epigenetic Diversity in Natural Animal Populations
3. Correlation between Epigenetic and Genetic Variation in Natural Animal Populations
4. Epigenetic Dynamics in Natural Animal Populations
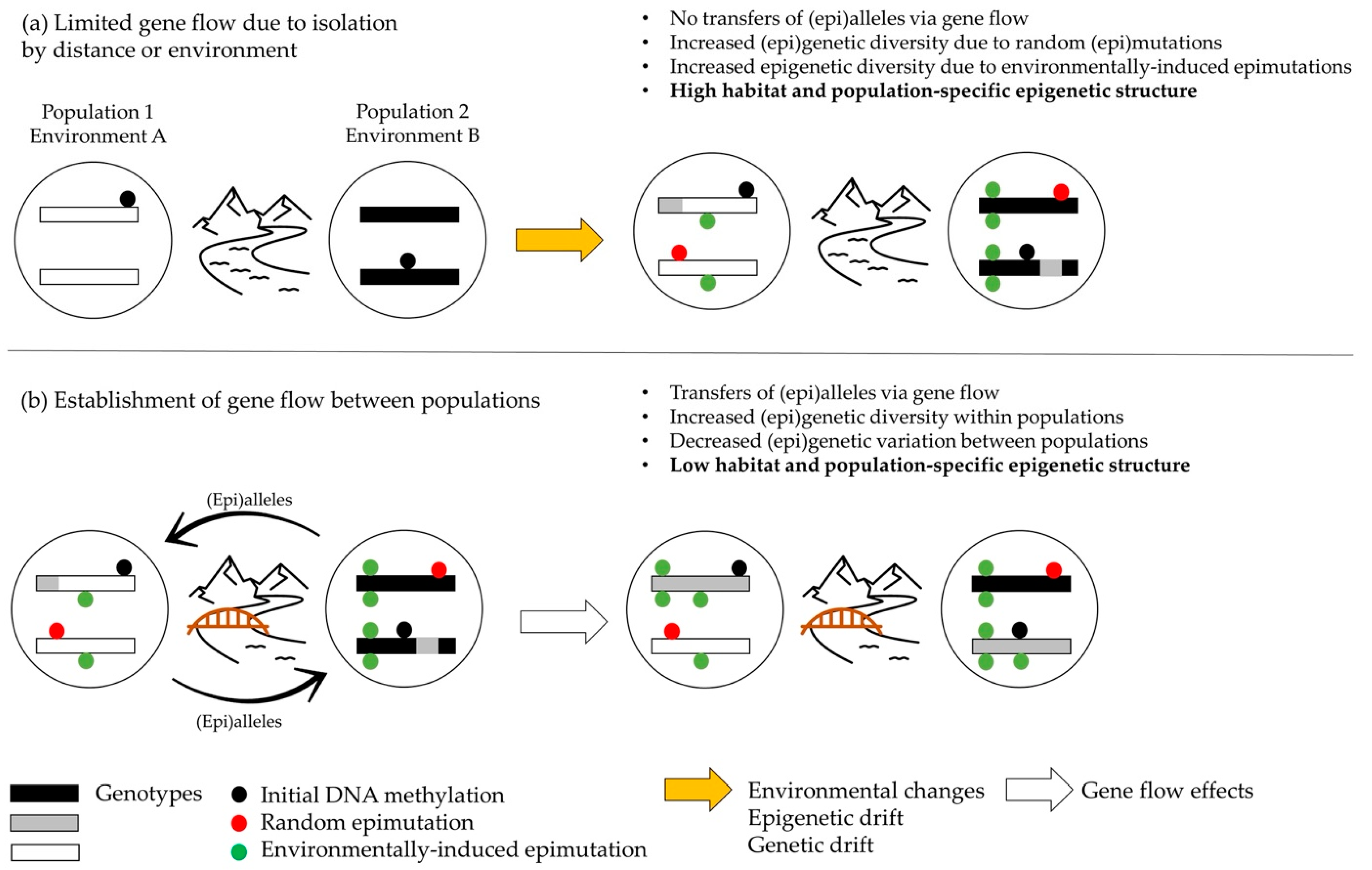
4.1. Geographical and Ecological Processes Acting on Both Epigenetic and Genetic Diversities
4.2. Ecological Processes Increasing Epigenetic Diversity
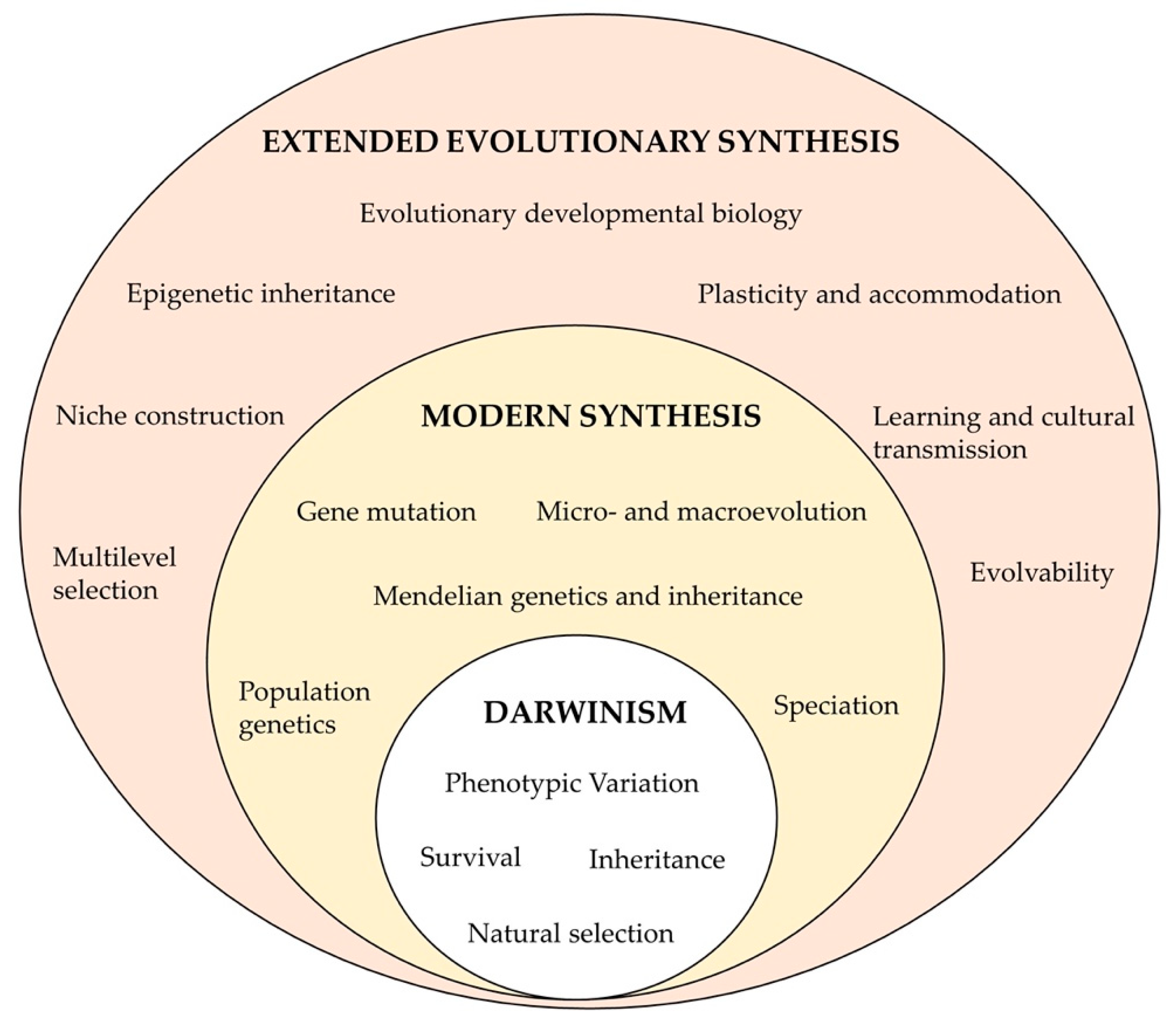
5. Epigenetic Variation as an Evolutionary Mechanism in Natural Populations
5.1. Epigenetics and Microevolution of Natural Animal Populations
5.2. Epigenetics and Macroevolution of Natural Animal Population
6. Future Research in Animal Population Epigenetics
7. Conclusions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Term | Definition |
|---|---|
| Epigenetic mark | Chemical modifications to DNA, RNA, or proteins that influence chromatin state and gene expression. It includes DNA methylation, noncoding RNAs, and protein modifications (e.g., acetylation, deacetylation, ubiquitination, and histone methylation). |
| Epiallele | Locus presenting distinct epigenetic profiles due to differences in methylation or chromatin states. |
| Epimutation | Heritable change in gene activity that is not associated with a DNA mutation, but rather with the gain or loss of DNA methylation or other heritable modifications of chromatin. |
| Single methylation polymorphism (SMP) | Spontaneous variation in DNA methylation at base-pair resolutions that are due to errors in the maintenance of methylation states. The rates of SMP formation is at least four orders of magnitude greater than genetic mutations. |
| Epigenetic reprogramming | Erasure and remodelling of epigenetic marks such as DNA methylation during embryo development. Its purposes include the erasure and reestablishment of parental genomic imprints in germ cells, the erasure of epimutations, and the correct development of the embryo through the generation of totipotent or multipotent cells. |
| Epigenetic variation | Variation in epialleles which is studied among and/or within populations. |
| Epigenetic diversity | The amount of epigenetic variation within a population. |
| Transgenerational epigenetic inheritance (TEI) | Stable inheritance of epigenetic marks across multiple generations. |
| Epigenetic divergence | The process in which two or more populations of an ancestral species accumulate independent epimutations through time. |
| Epigenetic drift | Gradual changes in the epigenome that is due to random epimutations. This neutral process is not directional as it creates both hyper- and hypomethylation. |
| CpG island | Short CpG-rich region of the genome characterized by at least 500 bp of DNA with a GC content ≥ 55%. |
| Epigenetic potential | The genomic capacity for environmentally induced phenotypic change (i.e., plasticity) via epigenetic modifications. |
| Phenotypic plasticity | Any change in an organism’s phenotype in response to an environmental signal. |
| Neo-Lamarckism or Lamarckian inheritance | A theory of evolution based on the principle of soft inheritance, which refers to the inheritance of variations that are the result of non-genetic effects. It includes inheritance coming from evolutionary developmental biology, epigenetics, niche construction, and learning and cultural transmission. This theory is part of the extended evolutionary synthesis. |
| Extended evolutionary synthesis | A set of evolutionary theories including the modern synthesis (combination of Darwinian view of evolutionary change and Mendelian genetics) and soft inheritance (or Lamarckian inheritance). This theory is still under debate. |
| Technique | Description, Advantages, and Limitations |
|---|---|
| Methylation-sensitive amplified polymorphism (MSAP) | Modified from the amplified fragment length polymorphism (AFLP) technique, MSAP uses EcoRI (rare cutter) with either one or two methylation-sensitive isoschizomer restriction enzymes, HpaII and MspI (frequent-cutter), which recognize the same restriction site (5′-CCGG-3′) but have different cytosine methylation sensitivities. For each sample, MSAP analysis is performed using both EcoRI/HpaII- and EcoRI/MspI-digested samples. The resulting DNA fragments are ligated with linkers and PCR amplified. Such amplification produces a reduced population of fragments that are separated in denaturing polyacrylamide gels in order to compare the respective band patterns. This technique is useful for non-model species as it does not require a reference genome. It is one of the most commonly used methods for assessing DNA methylation changes in plants. However, the main disadvantage of MSAP is that it can only detect methylation on 5′-CCGG-3′. |
| Methylated DNA immunoprecipitation (MeDIP)-Seq | MeDIP is an enrichment-based purification technique that involves antibodies directed against mC or mCG to precipitate methylated DNA fragments. Differential DNA methylation regions are identified by comparing the coverage between groups of interest. Combining MeDIP with next-generation sequencing, it provides methylomes at typically 100-bp to 300-bp resolution. With the appropriate antibody, MeDIP is also able to detect hmC. MeDIP limitations include antibody quality and cross-reactivity, and relatively low-resolution level in comparison with bisulfite sequencing methods. |
| Reduced representation bisulfite sequencing (RRBS) | RRBS relies on digestion of genomic DNA with the enzyme MspI, which produces DNA fragments that begin and/or end with an informative CpG site (CpG-enriched genomic regions). Then, genomic DNA is treated with sodium bisulfite, which leaves methylated cytosines intact but converts unmethylated cytosines to uracil (and ultimately thymine after PCR). Amplification fragments are sequenced, allowing for the identification of methylated cytosines. This is an efficient and high-throughput technique due to its high definition since it produces genome-wide methylation profiles with single-nucleotide resolution. Compared to WGBS, it allows one to investigate larger numbers of individuals as it is more cost-effective, but it only provides limited genome coverage (5–10%) and is CpG island and promoter region-centric. |
| Whole-genome bisulfite sequencing (WGBS | WGBS combines the use of sodium bisulfite treatment and high-throughput DNA sequencing to produce genome-wide methylation profiles with single-nucleotide resolution. Unlike RRBS, it estimates all cytosines methylation levels (including CpG and non-CpG) across the genome, rather than CpG enriched genomic regions. This method is capable of testing approximately 90% of all cytosines in genomes studied to date but is cost prohibitive to sequence large numbers of individual samples. |
References
- Mameli, M. Nongenetic Selection and Nongenetic Inheritance. Br. J. Philos. Sci. 2004, 55, 35–71. [Google Scholar] [CrossRef]
- Danchin, É.; Charmantier, A.; Champagne, F.A.; Mesoudi, A.; Pujol, B.; Blanchet, S. Beyond DNA: Integrating Inclusive Inheritance into an Extended Theory of Evolution. Nat. Rev. Genet. 2011, 12, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.D.; Norris, M.L.; Surani, M.A. Epigenetic Control of Transgene Expression and Imprinting by Genotype-Specific Modifiers. Cell 1990, 61, 853–861. [Google Scholar] [CrossRef]
- Miko, I. Phenotype Variability: Penetrance and Expressivity. Nat. Educ. 2008, 1, 137. [Google Scholar]
- Youngson, N.A.; Whitelaw, E. Transgenerational Epigenetic Effects. Annu. Rev. Genom. Hum. Genet. 2008, 9, 233–257. [Google Scholar] [CrossRef] [PubMed]
- Nicoglou, A.; Merlin, F. Epigenetics: A Way to Bridge the Gap between Biological Fields. Stud. Hist. Philos. Sci. Part C Stud. Hist. Philos. Biol. Biomed. Sci. 2017, 66, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Angers, B.; Castonguay, E.; Massicotte, R. Environmentally Induced Phenotypes and DNA Methylation: How to Deal with Unpredictable Conditions until the next Generation and After. Mol. Ecol. 2010, 19, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Greally, J.M. Population Epigenetics. Curr. Opin. Syst. Biol. 2017, 1, 84–89. [Google Scholar] [CrossRef]
- Verhoeven, K.J.F.; Preite, V. Epigenetic Variation in Asexually Reproducing Organisms. Evolution 2014, 68, 644–655. [Google Scholar] [CrossRef]
- West-Eberhard, M.J. Developmental Plasticity and Evolution; Oxford University Press: New York, NY, USA, 2003; ISBN 978-0-19-802856-7. [Google Scholar]
- West-Eberhard, M.J. Developmental Plasticity and the Origin of Species Differences. Proc. Natl. Acad. Sci. USA 2005, 102, 6543–6549. [Google Scholar] [CrossRef]
- Robinson, B.W.; Dukas, R. The Influence of Phenotypic Modifications on Evolution: The Baldwin Effect and Modern Perspectives. Oikos 1999, 85, 582. [Google Scholar] [CrossRef] [Green Version]
- Grether, G.F. Environmental Change, Phenotypic Plasticity, and Genetic Compensation. Am. Nat. 2005, 166, E115–E123. [Google Scholar] [CrossRef] [PubMed]
- Jablonka, E.; Lamb, M.J. The Inheritance of Acquired Epigenetic Variations. J. Theor. Biol. 1989, 139, 69–83. [Google Scholar] [CrossRef]
- West-Eberhard, M.J. Alternative Adaptations, Speciation, and Phylogeny (A Review). Proc. Natl. Acad. Sci. USA 1986, 83, 1388–1392. [Google Scholar] [CrossRef]
- Richards, E.J. Natural Epigenetic Variation in Plant Species: A View from the Field. Curr. Opin. Plant Biol. 2011, 14, 204–209. [Google Scholar] [CrossRef]
- Vogt, G. Epigenetic Variation in Animal Populations: Sources, Extent, Phenotypic Implications, and Ecological and Evolutionary Relevance. J. Biosci. 2021, 46, 24. [Google Scholar] [CrossRef]
- Burggren, W. Epigenetic Inheritance and Its Role in Evolutionary Biology: Re-Evaluation and New Perspectives. Biology 2016, 5, 24. [Google Scholar] [CrossRef]
- Kumar, D.; Thakur, M.K. Effect of Perinatal Exposure to Bisphenol-A on DNA Methylation and Histone Acetylation in Cerebral Cortex and Hippocampus of Postnatal Male Mice. J. Toxicol. Sci. 2017, 42, 281–289. [Google Scholar] [CrossRef]
- Nguyen, T.; Li, G.E.; Chen, H.; Cranfield, C.G.; McGrath, K.C.; Gorrie, C.A. Maternal E-Cigarette Exposure Results in Cognitive and Epigenetic Alterations in Offspring in a Mouse Model. Chem. Res. Toxicol. 2018, 31, 601–611. [Google Scholar] [CrossRef]
- Schmitz, R.J.; Schultz, M.D.; Lewsey, M.G.; O’Malley, R.C.; Urich, M.A.; Libiger, O.; Schork, N.J.; Ecker, J.R. Transgenerational Epigenetic Instability Is a Source of Novel Methylation Variants. Science 2011, 334, 369–373. [Google Scholar] [CrossRef]
- van der Graaf, A.; Wardenaar, R.; Neumann, D.A.; Taudt, A.; Shaw, R.G.; Jansen, R.C.; Schmitz, R.J.; Colomé-Tatché, M.; Johannes, F. Rate, Spectrum, and Evolutionary Dynamics of Spontaneous Epimutations. Proc. Natl. Acad. Sci. USA 2015, 112, 6676–6681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manikkam, M.; Guerrero-Bosagna, C.; Tracey, R.; Haque, M.M.; Skinner, M.K. Transgenerational Actions of Environmental Compounds on Reproductive Disease and Identification of Epigenetic Biomarkers of Ancestral Exposures. PLoS ONE 2012, 7, e31901. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Bosagna, C.; Morisson, M.; Liaubet, L.; Rodenburg, T.B.; de Haas, E.N.; Košťál, Ľ.; Pitel, F. Transgenerational Epigenetic Inheritance in Birds. Environ. Epigenetics 2018, 4, dvy008. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, R.K.; vom Saal, F.S.; Tillitt, D.E. Transgenerational Effects from Early Developmental Exposures to Bisphenol A or 17α-Ethinylestradiol in Medaka, Oryzias Latipes. Sci. Rep. 2015, 5, 9303. [Google Scholar] [CrossRef] [PubMed]
- Seong, K.-H.; Li, D.; Shimizu, H.; Nakamura, R.; Ishii, S. Inheritance of Stress-Induced, ATF-2-Dependent Epigenetic Change. Cell 2011, 145, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Liew, Y.J.; Howells, E.J.; Wang, X.; Michell, C.T.; Burt, J.A.; Idaghdour, Y.; Aranda, M. Intergenerational Epigenetic Inheritance in Reef-Building Corals. Nat. Clim. Change 2020, 10, 254–259. [Google Scholar] [CrossRef]
- Feil, R.; Fraga, M.F. Epigenetics and the Environment: Emerging Patterns and Implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef]
- Casier, K.; Boivin, A.; Carré, C.; Teysset, L. Environmentally-Induced Transgenerational Epigenetic Inheritance: Implication of PIWI Interacting RNAs. Cells 2019, 8, 1108. [Google Scholar] [CrossRef]
- Miryeganeh, M.; Saze, H. Epigenetic Inheritance and Plant Evolution. Popul. Ecol. 2019, 62, 17–27. [Google Scholar] [CrossRef]
- Thiebaut, F.; Hemerly, A.S.; Ferreira, P.C.G. A Role for Epigenetic Regulation in the Adaptation and Stress Responses of Non-Model Plants. Front. Plant Sci. 2019, 10, 246. [Google Scholar] [CrossRef]
- Blouin, M.S.; Thuillier, V.; Cooper, B.; Amarasinghe, V.; Cluzel, L.; Araki, H.; Grunau, C. No Evidence for Large Differences in Genomic Methylation between Wild and Hatchery Steelhead (Oncorhynchus Mykiss). Can. J. Fish. Aquat. Sci. 2010, 67, 217–224. [Google Scholar] [CrossRef]
- de Mendoza, A.; Lister, R.; Bogdanovic, O. Evolution of DNA Methylome Diversity in Eukaryotes. J. Mol. Biol. 2020, 432, 1687–1705. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-Wide Evolutionary Analysis of Eukaryotic DNA Methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef]
- Rauluseviciute, I.; Drabløs, F.; Rye, M.B. DNA Hypermethylation Associated with Upregulated Gene Expression in Prostate Cancer Demonstrates the Diversity of Epigenetic Regulation. BMC Med. Genomics 2020, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Spainhour, J.C.; Lim, H.S.; Yi, S.V.; Qiu, P. Correlation Patterns Between DNA Methylation and Gene Expression in The Cancer Genome Atlas. Cancer Inf. 2019, 18, 117693511982877. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.-Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and Divergence of Methylation Patterning in Plants and Animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA Methylation Landscapes: Provocative Insights from Epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Hon, G.C.; Rajagopal, N.; Shen, Y.; McCleary, D.F.; Yue, F.; Dang, M.D.; Ren, B. Epigenetic Memory at Embryonic Enhancers Identified in DNA Methylation Maps from Adult Mouse Tissues. Nat. Genet. 2013, 45, 1198–1206. [Google Scholar] [CrossRef]
- de Mendoza, A.; Hatleberg, W.L.; Pang, K.; Leininger, S.; Bogdanovic, O.; Pflueger, J.; Buckberry, S.; Technau, U.; Hejnol, A.; Adamska, M.; et al. Convergent Evolution of a Vertebrate-like Methylome in a Marine Sponge. Nat. Ecol. Evol. 2019, 3, 1464–1473. [Google Scholar] [CrossRef]
- Potok, M.E.; Nix, D.A.; Parnell, T.J.; Cairns, B.R. Reprogramming the Maternal Zebrafish Genome after Fertilization to Match the Paternal Methylation Pattern. Cell 2013, 153, 759–772. [Google Scholar] [CrossRef]
- Rae, P.M.M.; Steele, R.E. Absence of Cytosine Methylation at C-C-G-G and G-C-G-C Sites in the RDNA Coding Regions and Intervening Sequences of Drosophila and the RDNA of Other Higher Insects. Nucl. Acids. Res. 1979, 6, 2987–2995. [Google Scholar] [CrossRef] [PubMed]
- Simpson, V.J.; Johnson, T.E.; Hammen, R.F. Caenorhabditis Elegans DNA Does Not Contain 5-Methylcytosine at Any Time during Development or Aging. Nucl. Acids. Res. 1986, 14, 6711–6719. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, G.; Li, C.; Zhang, J.; Wang, Q.; Simmons, D.K.; Chen, X.; Wijesena, N.; Zhu, W.; Wang, Z.; et al. Evolutionary Transition between Invertebrates and Vertebrates via Methylation Reprogramming in Embryogenesis. Natl. Sci. Rev. 2019, 6, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Barrett, R.D.H. Epigenetics in Natural Animal Populations. J. Evol. Biol. 2017, 30, 1612–1632. [Google Scholar] [CrossRef]
- Schmitz, R.J.; Schultz, M.D.; Urich, M.A.; Nery, J.R.; Pelizzola, M.; Libiger, O.; Alix, A.; McCosh, R.B.; Chen, H.; Schork, N.J.; et al. Patterns of Population Epigenomic Diversity. Nature 2013, 495, 193–198. [Google Scholar] [CrossRef]
- Biwer, C.; Kawam, B.; Chapelle, V.; Silvestre, F. The Role of Stochasticity in the Origin of Epigenetic Variation in Animal Populations. Integr. Comp. Biol. 2020, 60, 1544–1557. [Google Scholar] [CrossRef]
- Oey, H.; Whitelaw, E. On the Meaning of the Word ‘Epimutation’. Trends Genet. 2014, 30, 519–520. [Google Scholar] [CrossRef]
- Ardura, A.; Zaiko, A.; Morán, P.; Planes, S.; Garcia-Vazquez, E. Epigenetic Signatures of Invasive Status in Populations of Marine Invertebrates. Sci. Rep. 2017, 7, 42193. [Google Scholar] [CrossRef]
- Baldanzi, S.; Watson, R.; McQuaid, C.D.; Gouws, G.; Porri, F. Epigenetic Variation among Natural Populations of the South African Sandhopper Talorchestia Capensis. Evol. Ecol. 2017, 31, 77–91. [Google Scholar] [CrossRef]
- Whitaker, J.M.; Welsh, A.B.; Hondorp, D.W.; Boase, J.C.; Merovich, G.T.; Welsh, S.; Krueger, C. Variation in DNA Methylation Is Associated with Migratory Phenotypes of Lake Sturgeon Acipenser fulvescens in the St. Clair River, MI, USA. J. Fish Biol. 2018, 93, 942–951. [Google Scholar] [CrossRef]
- Gavery, M.R.; Nichols, K.M.; Goetz, G.W.; Middleton, M.A.; Swanson, P. Characterization of Genetic and Epigenetic Variation in Sperm and Red Blood Cells from Adult Hatchery and Natural-Origin Steelhead, Oncorhynchus Mykiss. G3 Genes|Genomes|Genet. 2018, 8, 3723–3736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.M.; Kelly, M.W. Population Epigenetic Divergence Exceeds Genetic Divergence in the Eastern Oyster Crassostrea virginica in the Northern Gulf of Mexico. Evol. Appl. 2020, 13, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Wogan, G.O.U.; Yuan, M.L.; Mahler, D.L.; Wang, I.J. Genome-wide Epigenetic Isolation by Environment in a Widespread Anolis Lizard. Mol. Ecol. 2020, 29, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Watson, H.; Powell, D.; Salmón, P.; Jacobs, A.; Isaksson, C. Urbanization Is Associated with Modifications in DNA Methylation in a Small Passerine Bird. Evol. Appl. 2021, 14, 85–98. [Google Scholar] [CrossRef] [PubMed]
- McNew, S.M.; Beck, D.; Sadler-Riggleman, I.; Knutie, S.A.; Koop, J.A.H.; Clayton, D.H.; Skinner, M.K. Epigenetic Variation between Urban and Rural Populations of Darwin’s Finches. BMC Evol. Biol. 2017, 17, 183. [Google Scholar] [CrossRef] [PubMed]
- Thorson, J.L.M.; Smithson, M.; Sadler-Riggleman, I.; Beck, D.; Dybdahl, M.; Skinner, M.K. Regional Epigenetic Variation in Asexual Snail Populations among Urban and Rural Lakes. Environ. Epigenetics 2019, 5, dvz020. [Google Scholar] [CrossRef]
- Wang, X.; Li, A.; Wang, W.; Zhang, G.; Li, L. Direct and Heritable Effects of Natural Tidal Environments on DNA Methylation in Pacific Oysters (Crassostrea Gigas). Environ. Res. 2021, 197, 111058. [Google Scholar] [CrossRef]
- Flatscher, R.; Frajman, B.; Schönswetter, P.; Paun, O. Environmental Heterogeneity and Phenotypic Divergence: Can Heritable Epigenetic Variation Aid Speciation? Genet. Res. Int. 2012, 2012, 1–9. [Google Scholar] [CrossRef]
- Smith, T.A.; Martin, M.D.; Nguyen, M.; Mendelson, T.C. Epigenetic Divergence as a Potential First Step in Darter Speciation. Mol. Ecol. 2016, 25, 1883–1894. [Google Scholar] [CrossRef]
- Lira-Medeiros, C.F.; Parisod, C.; Fernandes, R.A.; Mata, C.S.; Cardoso, M.A.; Ferreira, P.C.G. Epigenetic Variation in Mangrove Plants Occurring in Contrasting Natural Environment. PLoS ONE 2010, 5, e10326. [Google Scholar] [CrossRef]
- Medrano, M.; Herrera, C.M.; Bazaga, P. Epigenetic Variation Predicts Regional and Local Intraspecific Functional Diversity in a Perennial Herb. Mol. Ecol. 2014, 23, 4926–4938. [Google Scholar] [CrossRef] [PubMed]
- Foust, C.M.; Preite, V.; Schrey, A.W.; Alvarez, M.; Robertson, M.H.; Verhoeven, K.J.F.; Richards, C.L. Genetic and Epigenetic Differences Associated with Environmental Gradients in Replicate Populations of Two Salt Marsh Perennials. Mol. Ecol. 2016, 25, 1639–1652. [Google Scholar] [CrossRef] [PubMed]
- Morán, P.; Pérez-Figueroa, A. Methylation Changes Associated with Early Maturation Stages in the Atlantic Salmon. BMC Genet. 2011, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Skinner, M.K.; Gurerrero-Bosagna, C.; Haque, M.M.; Nilsson, E.E.; Koop, J.A.H.; Knutie, S.A.; Clayton, D.H. Epigenetics and the Evolution of Darwin’s Finches. Genome Biol. Evol. 2014, 6, 1972–1989. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Sun, K.; Jiang, T.; Feng, J. Natural Epigenetic Variation in Bats and Its Role in Evolution. J. Exp. Biol. 2015, 218, 100–106. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Q.; Kong, L.; Yu, H. Epigenetic Variation of Wild Populations of the Pacific Oyster Crassostrea Gigas Determined by Methylation-Sensitive Amplified Polymorphism Analysis. Fish Sci. 2018, 84, 61–70. [Google Scholar] [CrossRef]
- Vernaz, G.; Malinsky, M.; Svardal, H.; Du, M.; Tyers, A.M.; Santos, M.E.; Durbin, R.; Genner, M.J.; Turner, G.F.; Miska, E.A. Mapping Epigenetic Divergence in the Massive Radiation of Lake Malawi Cichlid Fishes. Nat. Commun. 2021, 12, 5870. [Google Scholar] [CrossRef]
- Massicotte, R.; Whitelaw, E.; Angers, B. DNA Methylation: A Source of Random Variation in Natural Populations. Epigenetics 2011, 6, 421–427. [Google Scholar] [CrossRef]
- Massicotte, R.; Angers, B. General-Purpose Genotype or How Epigenetics Extend the Flexibility of a Genotype. Genet. Res. Int. 2012, 2012, 317175. [Google Scholar] [CrossRef]
- Thorson, J.L.M.; Smithson, M.; Beck, D.; Sadler-Riggleman, I.; Nilsson, E.; Dybdahl, M.; Skinner, M.K. Epigenetics and Adaptive Phenotypic Variation between Habitats in an Asexual Snail. Sci. Rep. 2017, 7, 14139. [Google Scholar] [CrossRef]
- Berbel-Filho, W.M.; Rodríguez-Barreto, D.; Berry, N.; Garcia De Leaniz, C.; Consuegra, S. Contrasting DNA Methylation Responses of Inbred Fish Lines to Different Rearing Environments. Epigenetics 2019, 14, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Richards, E.J. Inherited Epigenetic Variation—Revisiting Soft Inheritance. Nat. Rev. Genet. 2006, 7, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, E.L.; Schrey, A.; Andrew, S.C.; Ragsdale, A.; Griffith, S.C. Epigenetic and Genetic Variation among Three Separate Introductions of the House Sparrow (Passer domesticus) into Australia. R. Soc. Open Sci. 2018, 5, 172185. [Google Scholar] [CrossRef] [PubMed]
- Fargeot, L.; Loot, G.; Prunier, J.G.; Rey, O.; Veyssière, C.; Blanchet, S. Patterns of Epigenetic Diversity in Two Sympatric Fish Species: Genetic vs. Environmental Determinants. Genes 2021, 12, 107. [Google Scholar] [CrossRef] [PubMed]
- McRae, A.F.; Powell, J.E.; Henders, A.K.; Bowdler, L.; Hemani, G.; Shah, S.; Painter, J.N.; Martin, N.G.; Visscher, P.M.; Montgomery, G.W. Contribution of Genetic Variation to Transgenerational Inheritance of DNA Methylation. Genome Biol. 2014, 15, R73. [Google Scholar] [CrossRef]
- Carja, O.; MacIsaac, J.L.; Mah, S.M.; Henn, B.M.; Kobor, M.S.; Feldman, M.W.; Fraser, H.B. Worldwide Patterns of Human Epigenetic Variation. Nat. Ecol. Evol. 2017, 1, 1577–1583. [Google Scholar] [CrossRef]
- Orozco, L.D.; Morselli, M.; Rubbi, L.; Guo, W.; Go, J.; Shi, H.; Lopez, D.; Furlotte, N.A.; Bennett, B.J.; Farber, C.R.; et al. Epigenome-Wide Association of Liver Methylation Patterns and Complex Metabolic Traits in Mice. Cell Metab. 2015, 21, 905–917. [Google Scholar] [CrossRef]
- Liebl, A.L.; Schrey, A.W.; Richards, C.L.; Martin, L.B. Patterns of DNA Methylation Throughout a Range Expansion of an Introduced Songbird. Integr. Comp. Biol. 2013, 53, 351–358. [Google Scholar] [CrossRef]
- Leung, C.; Breton, S.; Angers, B. Facing Environmental Predictability with Different Sources of Epigenetic Variation. Ecol. Evol. 2016, 6, 5234–5245. [Google Scholar] [CrossRef]
- Wenzel, M.A.; Piertney, S.B. Fine-Scale Population Epigenetic Structure in Relation to Gastrointestinal Parasite Load in Red Grouse (Lagopus lagopus scotica). Mol. Ecol. 2014, 23, 4256–4273. [Google Scholar] [CrossRef]
- Venney, C.J.; Sutherland, B.J.G.; Beacham, T.D.; Heath, D.D. Population Differences in Chinook Salmon (Oncorhynchus tshawytscha) DNA Methylation: Genetic Drift and Environmental Factors. Ecol. Evol. 2021, 11, 6846–6861. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Martienssen, R. Transposable Elements and the Epigenetic Regulation of the Genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Elbarbary, R.A.; Lucas, B.A.; Maquat, L.E. Retrotransposons as Regulators of Gene Expression. Science 2016, 351, aac7247. [Google Scholar] [CrossRef] [PubMed]
- Schauer, S.N.; Carreira, P.E.; Shukla, R.; Gerhardt, D.J.; Gerdes, P.; Sanchez-Luque, F.J.; Nicoli, P.; Kindlova, M.; Ghisletti, S.; Santos, A.D.; et al. L1 Retrotransposition Is a Common Feature of Mammalian Hepatocarcinogenesis. Genome Res. 2018, 28, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ali, M.; Zhou, Q. Establishment and Evolution of Heterochromatin. Ann. N. Y. Acad. Sci. 2020, 1476, 59–77. [Google Scholar] [CrossRef]
- Platt, A.; Gugger, P.F.; Pellegrini, M.; Sork, V.L. Genome-Wide Signature of Local Adaptation Linked to Variable CpG Methylation in Oak Populations. Mol. Ecol. 2015, 24, 3823–3830. [Google Scholar] [CrossRef]
- Macia, A.; Muñoz-Lopez, M.; Cortes, J.L.; Hastings, R.K.; Morell, S.; Lucena-Aguilar, G.; Marchal, J.A.; Badge, R.M.; Garcia-Perez, J.L. Epigenetic Control of Retrotransposon Expression in Human Embryonic Stem Cells. Mol. Cell. Biol. 2011, 31, 300–316. [Google Scholar] [CrossRef]
- Tang, M.-H.; Varadan, V.; Kamalakaran, S.; Zhang, M.Q.; Dimitrova, N.; Hicks, J. Major Chromosomal Breakpoint Intervals in Breast Cancer Co-Localize with Differentially Methylated Regions. Front. Oncol. 2012, 2, 197. [Google Scholar] [CrossRef]
- Makova, K.D.; Hardison, R.C. The Effects of Chromatin Organization on Variation in Mutation Rates in the Genome. Nat. Rev. Genet. 2015, 16, 213–223. [Google Scholar] [CrossRef]
- Duncan, B.K.; Miller, J.H. Mutagenic Deamination of Cytosine Residues in DNA. Nature 1980, 287, 560–561. [Google Scholar] [CrossRef]
- Britten, R.J.; Baron, W.F.; Stout, D.B.; Davidson, E.H. Sources and Evolution of Human Alu Repeated Sequences. Proc. Natl. Acad. Sci. USA 1988, 85, 4770–4774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilvitis, H.J.; Hanson, H.; Schrey, A.W.; Martin, L.B. Epigenetic Potential as a Mechanism of Phenotypic Plasticity in Vertebrate Range Expansions. Integr. Comp. Biol. 2017, 57, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Isolation by distance. Genetics 1943, 28, 114–138. [Google Scholar] [CrossRef] [PubMed]
- Herrera, C.M.; Medrano, M.; Bazaga, P. Comparative Epigenetic and Genetic Spatial Structure of the Perennial Herb Helleborus foetidus: Isolation by Environment, Isolation by Distance, and Functional Trait Divergence. Am. J. Bot. 2017, 104, 1195–1204. [Google Scholar] [CrossRef]
- Wang, I.J.; Bradburd, G.S. Isolation by Environment. Mol. Ecol. 2014, 23, 5649–5662. [Google Scholar] [CrossRef]
- Mendelson, T.C.; Imhoff, V.E.; Venditti, J.J. The accumulation of reproductive barriers during speciation: Postmating barriers in two behaviorally isolated species of darters (Percidae: Etheostoma). Evolution 2007, 61, 2596–2606. [Google Scholar] [CrossRef]
- Liu, S.; Sun, K.; Jiang, T.; Ho, J.P.; Liu, B.; Feng, J. Natural Epigenetic Variation in the Female Great Roundleaf Bat (Hipposideros armiger) Populations. Mol. Genet. Genom. 2012, 287, 643–650. [Google Scholar] [CrossRef]
- Becker, C.; Hagmann, J.; Müller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous Epigenetic Variation in the Arabidopsis Thaliana Methylome. Nature 2011, 480, 245–249. [Google Scholar] [CrossRef]
- Yates, P.A.; Burman, R.; Simpson, J.; Ponomoreva, O.N.; Thayer, M.J.; Turker, M.S. Silencing of Mouse Aprt Is a Gradual Process in Differentiated Cells. Mol. Cell. Biol. 2003, 23, 4461–4470. [Google Scholar] [CrossRef]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of Spontaneous Mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [CrossRef]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledó, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Weigel, D.; Lynch, M. The Rate and Molecular Spectrum of Spontaneous Mutations in Arabidopsis thaliana. Science 2010, 327, 92–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashapkin, V.V.; Kutueva, L.I.; Vanyushin, B.F. Epigenetic Clock: Just a Convenient Marker or an Active Driver of Aging? In Reviews on Biomarker Studies in Aging and Anti-Aging Research; Guest, P.C., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2019; Volume 1178, pp. 175–206. ISBN 978-3-030-25649-4. [Google Scholar]
- Hernando-Herraez, I.; Evano, B.; Stubbs, T.; Commere, P.-H.; Jan Bonder, M.; Clark, S.; Andrews, S.; Tajbakhsh, S.; Reik, W. Ageing Affects DNA Methylation Drift and Transcriptional Cell-to-Cell Variability in Mouse Muscle Stem Cells. Nat. Commun. 2019, 10, 4361. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.T.; Fei, Z.; Haghani, A.; Robeck, T.R.; Zoller, J.A.; Li, C.Z.; Zhang, J.; Ablaeva, J.; Adams, D.; Almunia, J.; et al. Universal DNA methylation age across mammalian tissues. BioRxiv 2021. [Google Scholar] [CrossRef]
- Mayne, B.; Korbie, D.; Kenchington, L.; Ezzy, B.; Berry, O.; Jarman, S. A DNA Methylation Age Predictor for Zebrafish. Aging 2020, 12, 24817–24835. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadi, D.; Piferrer, F. A Clockwork Fish: Age Prediction Using DNA Methylation-based Biomarkers in the European Seabass. Mol. Ecol. Resour. 2020, 20, 387–397. [Google Scholar] [CrossRef]
- Li, Y.; Tollefsbol, T.O. Age-Related Epigenetic Drift and Phenotypic Plasticity Loss: Implications in Prevention of Age-Related Human Diseases. Epigenomics 2016, 8, 1637–1651. [Google Scholar] [CrossRef]
- Guillette, L.J.; Parrott, B.B.; Nilsson, E.; Haque, M.M.; Skinner, M.K. Epigenetic Programming Alterations in Alligators from Environmentally Contaminated Lakes. Gen. Comp. Endocrinol. 2016, 238, 4–12. [Google Scholar] [CrossRef]
- Sheldon, C.C.; Conn, A.B.; Dennis, E.S.; Peacock, W.J. Different Regulatory Regions Are Required for the Vernalization-Induced Repression of FLOWERING LOCUS C and for the Epigenetic Maintenance of Repression. Plant Cell 2002, 14, 2527–2537. [Google Scholar] [CrossRef]
- Feil, R. Environmental and Nutritional Effects on the Epigenetic Regulation of Genes. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2006, 600, 46–57. [Google Scholar] [CrossRef]
- Meaney, M.J. Maternal Care, Gene Expression, and the Transmission of Individual Differences in Stress Reactivity Across Generations. Annu. Rev. Neurosci. 2001, 24, 1161–1192. [Google Scholar] [CrossRef]
- Voisin, A.-S.; Suarez Ulloa, V.; Stockwell, P.; Chatterjee, A.; Silvestre, F. Genome-Wide DNA Methylation of the Liver Reveals Delayed Effects of Early-Life Exposure to 17-α-Ethinylestradiol in the Self-Fertilizing Mangrove Rivulus. Epigenetics 2022, 17, 473–497. [Google Scholar] [CrossRef] [PubMed]
- Richards, E.J. Population Epigenetics. Curr. Opin. Genet. Dev. 2008, 18, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Artemov, A.V.; Mugue, N.S.; Rastorguev, S.M.; Zhenilo, S.; Mazur, A.M.; Tsygankova, S.V.; Boulygina, E.S.; Kaplun, D.; Nedoluzhko, A.V.; Medvedeva, Y.A.; et al. Genome-Wide DNA Methylation Profiling Reveals Epigenetic Adaptation of Stickleback to Marine and Freshwater Conditions. Mol. Biol. Evol. 2017, 34, 2203–2213. [Google Scholar] [CrossRef] [PubMed]
- Tatsch, A.; Proietti, M.; Zanini, R.; Fruet, P.; Secchi, E. Beyond Genetic Differences: Epigenetic Variation in Common Bottlenose Dolphins Tursiops Truncatus from Contrasting Marine Ecosystems. Mar. Ecol. Prog. Ser. 2021, 671, 219–233. [Google Scholar] [CrossRef]
- Lea, A.J.; Altmann, J.; Alberts, S.C.; Tung, J. Resource Base Influences Genome-Wide DNA Methylation Levels in Wild Baboons (Papio cynocephalus). Mol. Ecol. 2016, 25, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wuitchik, S.J.S.; Barry, T.N.; Jamniczky, H.A.; Rogers, S.M.; Barrett, R.D.H. Heritability of DNA Methylation in Threespine Stickleback (Gasterosteus aculeatus). Genetics 2021, 217, iyab001. [Google Scholar] [CrossRef]
- Mayr, E. The Growth of Biological Thought: Diversity, Evolution, and Inheritance; Harvard University Press: Cambridge, MA, USA, 1982; ISBN 978-0-674-36446-2. [Google Scholar]
- Pigliucci, M.; Müller, G.; Konrad Lorenz Institute for Evolution and Cognition Research (Eds.) Evolution, the Extended Synthesis; MIT Press: Cambridge, MA, USA, 2010; ISBN 978-0-262-51367-8. [Google Scholar]
- Skinner, M.K.; Guerrero-Bosagna, C.; Haque, M.M. Environmentally Induced Epigenetic Transgenerational Inheritance of Sperm Epimutations Promote Genetic Mutations. Epigenetics 2015, 10, 762–771. [Google Scholar] [CrossRef]
- Tigano, A.; Friesen, V. Genomics of Local Adaptation with Gene Flow. Mol. Ecol. 2016, 25, 2144–2164. [Google Scholar] [CrossRef]
- Xie, H.J.; Li, H.; Liu, D.; Dai, W.M.; He, J.Y.; Lin, S.; Duan, H.; Liu, L.L.; Chen, S.G.; Song, X.L.; et al. ICE1 Demethylation Drives the Range Expansion of a Plant Invader through Cold Tolerance Divergence. Mol. Ecol. 2015, 24, 835–850. [Google Scholar] [CrossRef]
- Vogt, G. Stochastic Developmental Variation, an Epigenetic Source of Phenotypic Diversity with Far-Reaching Biological Consequences. J. Biosci. 2015, 40, 159–204. [Google Scholar] [CrossRef]
- Simons, A.M. Modes of Response to Environmental Change and the Elusive Empirical Evidence for Bet Hedging. Proc. R. Soc. B. 2011, 278, 1601–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardura, A.; Clusa, L.; Zaiko, A.; Garcia-Vazquez, E.; Miralles, L. Stress Related Epigenetic Changes May Explain Opportunistic Success in Biological Invasions in Antipode Mussels. Sci. Rep. 2018, 8, 10793. [Google Scholar] [CrossRef] [PubMed]
- Schrey, A.W.; Coon, C.A.C.; Grispo, M.T.; Awad, M.; Imboma, T.; McCoy, E.D.; Mushinsky, H.R.; Richards, C.L.; Martin, L.B. Epigenetic Variation May Compensate for Decreased Genetic Variation with Introductions: A Case Study Using House Sparrows (Passer domesticus) on Two Continents. Genet. Res. Int. 2012, 2012, 979751. [Google Scholar] [CrossRef] [PubMed]
- Riyahi, S.; Vilatersana, R.; Schrey, A.W.; Ghorbani Node, H.; Aliabadian, M.; Senar, J.C. Natural Epigenetic Variation within and among Six Subspecies of the House Sparrow, Passer domesticus. J. Exp. Biol. 2017, 220, 4016–4023. [Google Scholar] [CrossRef]
- Young, R.L.; Badyaev, A.V. Evolution of Ontogeny: Linking Epigenetic Remodeling and Genetic Adaptation in Skeletal Structures. Integr. Comp. Biol. 2007, 47, 234–244. [Google Scholar] [CrossRef]
- Simpson, G.G. The Baldwin Effect. Evolution 1953, 7, 110. [Google Scholar] [CrossRef]
- Llamas, B.; Holland, M.L.; Chen, K.; Cropley, J.E.; Cooper, A.; Suter, C.M. High-Resolution Analysis of Cytosine Methylation in Ancient DNA. PLoS ONE 2012, 7, e30226. [Google Scholar] [CrossRef]
- Reik, W.; Dean, W.; Walter, J. Epigenetic Reprogramming in Mammalian Development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef]
- Mhanni, A.A.; McGowan, R.A. Global Changes in Genomic Methylation Levels during Early Development of the Zebrafish Embryo. Dev. Genes. Evol. 2004, 214, 412–417. [Google Scholar] [CrossRef]
- Fellous, A.; Labed-Veydert, T.; Locrel, M.; Voisin, A.-S.; Earley, R.L.; Silvestre, F. DNA Methylation in Adults and during Development of the Self-Fertilizing Mangrove Rivulus, Kryptolebias marmoratus. Ecol. Evol. 2018, 8, 6016–6033. [Google Scholar] [CrossRef]
- Wang, X.; Bhandari, R.K. DNA Methylation Dynamics during Epigenetic Reprogramming of Medaka Embryo. Epigenetics 2019, 14, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Costa, W.J.E.M.; Lima, S.M.Q.; Bartolette, R. Androdioecy in Kryptolebias Killifish and the Evolution of Self-Fertilizing Hermaphroditism. Biol. J. Linn. Soc. 2010, 99, 344–349. [Google Scholar] [CrossRef]
- Tatarenkov, A.; Earley, R.L.; Taylor, D.S.; Avise, J.C. Microevolutionary Distribution of Isogenicity in a Self-Fertilizing Fish (Kryptolebias marmoratus) in the Florida Keys. Integr. Comp. Biol. 2012, 52, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Mirbahai, L.; Chipman, J.K. Epigenetic Memory of Environmental Organisms: A Reflection of Lifetime Stressor Exposures. Mutat. Res./Genet. Toxicol. Environ. Mutagenesis 2014, 764–765, 10–17. [Google Scholar] [CrossRef]
- Carvan, M.J.; Kalluvila, T.A.; Klingler, R.H.; Larson, J.K.; Pickens, M.; Mora-Zamorano, F.X.; Connaughton, V.P.; Sadler-Riggleman, I.; Beck, D.; Skinner, M.K. Mercury-Induced Epigenetic Transgenerational Inheritance of Abnormal Neurobehavior Is Correlated with Sperm Epimutations in Zebrafish. PLoS ONE 2017, 12, e0176155. [Google Scholar] [CrossRef]
- Crotti, M.; Yohannes, E.; Winfield, I.J.; Lyle, A.A.; Adams, C.E.; Elmer, K.R. Rapid Adaptation through Genomic and Epigenomic Responses Following Translocations in an Endangered Salmonid. Evol. Appl. 2021, 14, 2470–2489. [Google Scholar] [CrossRef]
- Rey, O.; Eizaguirre, C.; Angers, B.; Baltazar-Soares, M.; Sagonas, K.; Prunier, J.G.; Blanchet, S. Linking Epigenetics and Biological Conservation: Towards a Conservation epigenetics Perspective. Funct. Ecol. 2020, 34, 414–427. [Google Scholar] [CrossRef]
- Ferguson-Smith, A.C. Genomic Imprinting: The Emergence of an Epigenetic Paradigm. Nat. Rev. Genet. 2011, 12, 565–575. [Google Scholar] [CrossRef]
- Lyon, M.F. Gene Action in the X-Chromosome of the Mouse (Mus musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef]
- Eckersley-Maslin, M.A.; Thybert, D.; Bergmann, J.H.; Marioni, J.C.; Flicek, P.; Spector, D.L. Random Monoallelic Gene Expression Increases upon Embryonic Stem Cell Differentiation. Dev. Cell 2014, 28, 351–365. [Google Scholar] [CrossRef]
- Jeffries, A.R.; Perfect, L.W.; Ledderose, J.; Schalkwyk, L.C.; Bray, N.J.; Mill, J.; Price, J. Stochastic Choice of Allelic Expression in Human Neural Stem Cells. Stem Cells 2012, 30, 1938–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinius, B.; Mold, J.E.; Ramsköld, D.; Deng, Q.; Johnsson, P.; Michaëlsson, J.; Frisén, J.; Sandberg, R. Analysis of Allelic Expression Patterns in Clonal Somatic Cells by Single-Cell RNA–Seq. Nat. Genet. 2016, 48, 1430–1435. [Google Scholar] [CrossRef] [PubMed]
- Akintola, A.D.; Crislip, Z.L.; Catania, J.M.; Chen, G.; Zimmer, W.E.; Burghardt, R.C.; Parrish, A.R. Promoter Methylation Is Associated with the Age-Dependent Loss of N-Cadherin in the Rat Kidney. Am. J. Physiol.-Ren. Physiol. 2008, 294, F170–F176. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.F.; Atzmon, G.; Gheorghe, C.; Liang, H.Q.; Lowes, C.; Greally, J.M.; Barzilai, N. Tissue-Specific Dysregulation of DNA Methylation in Aging: Tissue-Specific Epigenetic Dysregulation with Aging. Aging Cell 2010, 9, 506–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]



| Species | Genetic vs. Epigenetic Correlation | Epialleles Category | Ref. |
|---|---|---|---|
| Clonal fish (Chrosomus eos-neogaeus) | No significant correlation | Putatively pure or facilitated | [69] |
| Clonal fish (Chrosomus eos-neogaeus) | No significant correlation | Putatively pure or facilitated | [70] |
| Clonal fish (Chrosomus eos-neogaeus) | No significant correlation | Unknown | [80] |
| House sparrows (Passer domesticus) (Africa) | Significant negative correlation | Unknown | [79] |
| House sparrows (Passer domesticus) (Australia) | No significant correlation | Unknown | [74] |
| Red grouse (Lagopus lagopus scotica) | No significant correlation | Unknown | [81] |
| Bats (Rhinolophus pusillus, Hipposideros armiger and Miniopterus fuliginosus) | Significant positive correlation | Unknown | [66] |
| South African (Gansbaii) sandhopper (Talorchestia capensis) | No significant correlation | Putatively pure or facilitated | [50] |
| South African sandhopper (Talorchestia capensis) | Significant negative correlation | Putatively obligatory | [50] |
| Pacific oyster (Crassostrea gigas) | Significant positive correlation | Putatively obligatory | [67] |
| Crested anole (Anolis cristatellus) | Significant positive correlation | Putatively obligatory | [54] |
| Eastern oyster (Crassostrea virginica) | No significant correlation | Unknown | [53] |
| Fish (Gobio occitaniae) | Significant positive correlation | Unknown | [75] |
| Chinook salmon (Oncorhynchus tshawytscha) | No significant correlation | Unknown | [82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chapelle, V.; Silvestre, F. Population Epigenetics: The Extent of DNA Methylation Variation in Wild Animal Populations. Epigenomes 2022, 6, 31. https://doi.org/10.3390/epigenomes6040031
Chapelle V, Silvestre F. Population Epigenetics: The Extent of DNA Methylation Variation in Wild Animal Populations. Epigenomes. 2022; 6(4):31. https://doi.org/10.3390/epigenomes6040031
Chicago/Turabian StyleChapelle, Valentine, and Frédéric Silvestre. 2022. "Population Epigenetics: The Extent of DNA Methylation Variation in Wild Animal Populations" Epigenomes 6, no. 4: 31. https://doi.org/10.3390/epigenomes6040031