Quantitative Epigenetics: A New Avenue for Crop Improvement



Abstract
:1. Introduction
2. Epialleles (Natural and Mutagen Induced)
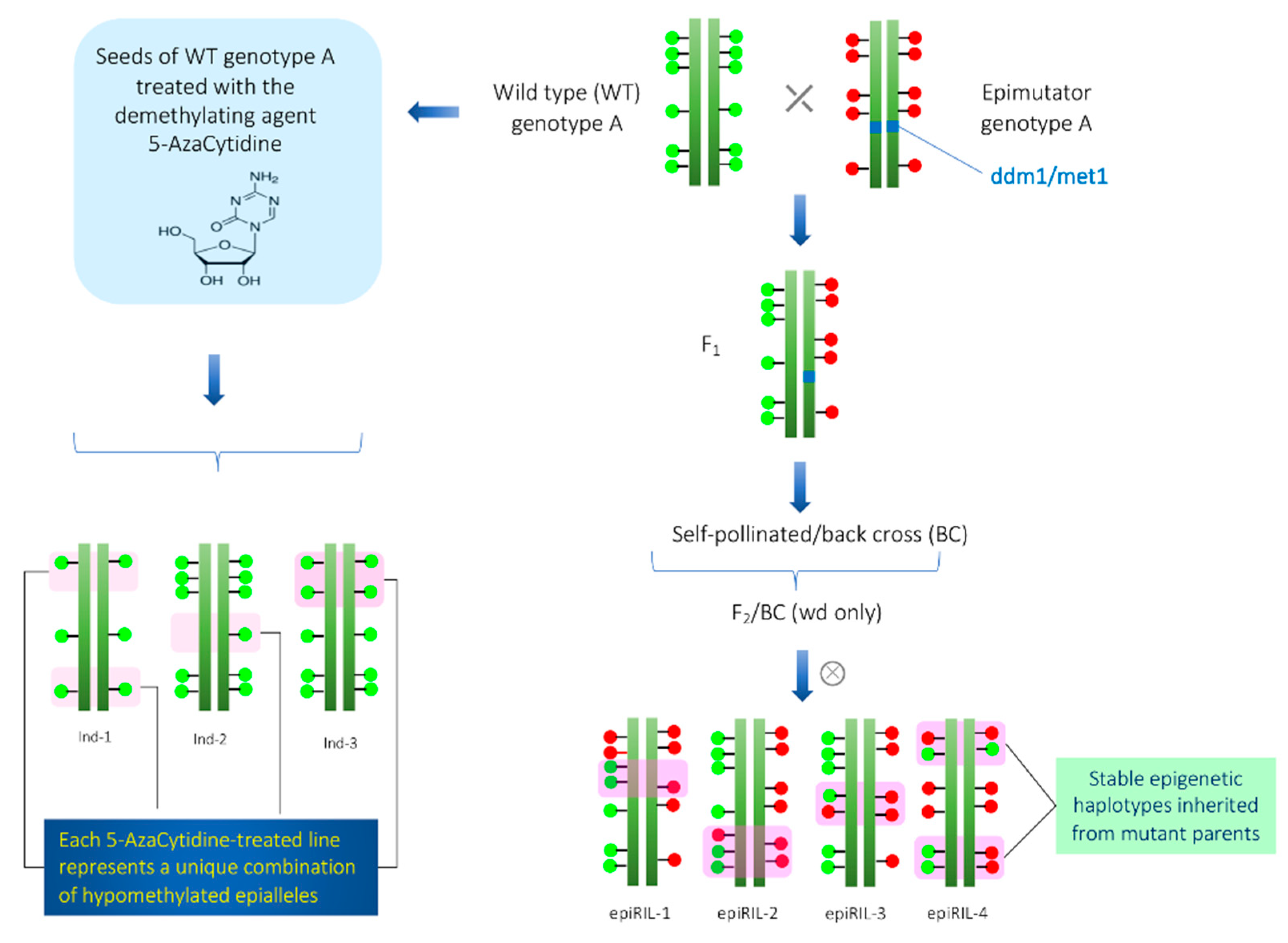
3. Epigenetic Recombinant Inbred Lines (epiRILs)
3.1. Persistence of Epigenetic Modification in the epiRILs
3.2. Phenotypic Variation and Stability in the epiRILs
3.3. Epigenetic Basis of Heterosis
4. Development of Epigenetically Modified Population by Chemical Agents
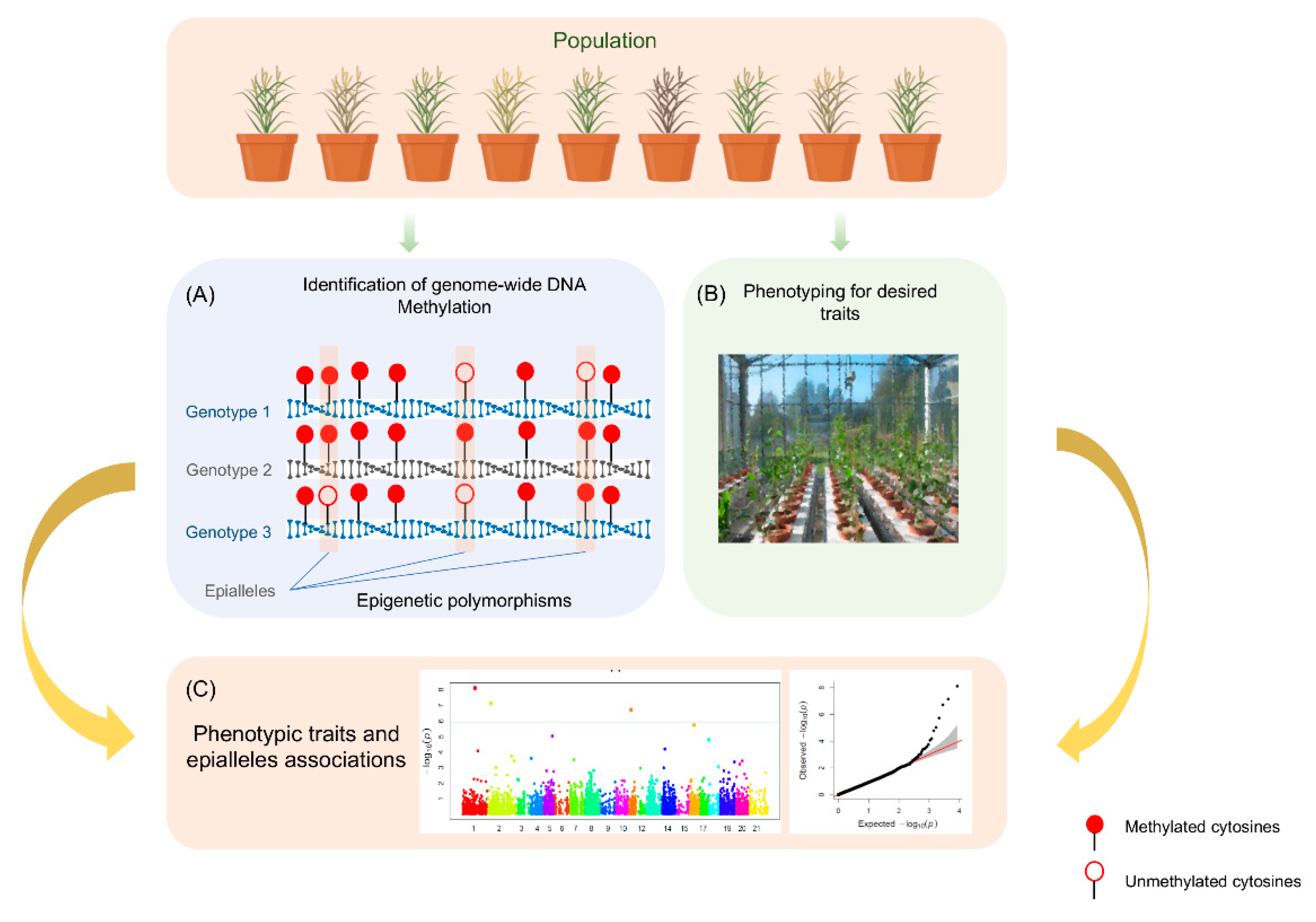
5. Development of User-Friendly Epigenetic Markers
6. Quantitative Epigenetic Models for Complex Trait
7. EpiQTLs and Epigenome-Wide Association Study (EWAS)
8. Epigenome Editing Using Site-Specific Nucleases
9. Conclusions and Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic reprogramming in plant and animal development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef] [Green Version]
- Simon, S.A.; Meyers, B.C. Small RNA-mediated epigenetic modifications in plants. Curr. Opin. Plant Biol. 2011, 14, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Ausin, I.; Johnson, L.M.; Vashisht, A.A.; Zhu, J.K.; Wohlschlegel, J.A.; Jacobsen, S.E. A protein complex required for polymerase V transcripts and RNA- directed DNA methylation in Arabidopsis. Curr. Biol. 2010, 20, 951–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Cao, X.; Aufsatz, W.; Zilberman, D.; Mette, M.F.; Huang, M.S.; Matzke, M.; Jacobsen, S.E. Role of the DRM and CMT3 methyltransferases in RNA-directed DNA methylation. Curr. Biol. 2003, 13, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- Gent, J.I.; Ellis, N.A.; Guo, L.; Harkess, A.E.; Yao, Y.; Zhang, X.; Dawe, R.K. CHH islands: De novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 2013, 23, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Saze, H.; Mittelsten Scheid, O.; Paszkowski, J. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat. Genet. 2003, 34, 65–69. [Google Scholar] [CrossRef]
- Zemach, A.; Kim, M.Y.; Hsieh, P.H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lang, Z.; Zhu, J. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Yong, W.; Hsu, F.; Chen, P. Profiling genome-wide DNA methylation. Epigenetics Chromatin 2016, 9, 26. [Google Scholar] [CrossRef] [Green Version]
- Kawakatsu, T.; Huang, S.C.; Jupe, F.; Sasaki, E.; Schmitz, R.J.; Urich, M.A.; Castanon, R.; Nery, J.R.; Barragan, C.; He, Y.; et al. Epigenomic Diversity in a Global Collection of Arabidopsis thaliana Accessions. Cell 2016, 166, 492–505. [Google Scholar] [CrossRef] [Green Version]
- Laird, P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Eichten, S.R.; Hermanson, P.J.; Springer, N.M. Inheritance patterns and stability of DNA methylation variation in maize near-isogenic lines. Genetics 2014, 196, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, L.J.; Quinton-Tulloch, M.; Olohan, L.; Price, J.; Hall, N.; Hall, A. A genome-wide survey of DNA methylation in hexaploid wheat. Genome Biol. 2015, 16, 273. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Singh, V.K.; Saxena, R.K.; Kale, S.M.; Li, Y.; Garg, V.; Meifang, T.; Khan, A.W.; Kim, K.D.; Chitikineni, A.; et al. Genome-wide analysis of epigenetic and transcriptional changes associated with heterosis in pigeonpea. Plant Biotechnol. J. 2020, 18, 1697–1710. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Shi, Y.; Chang, X.; Jing, S.; Zhang, Q.; You, C.; Yuan, H.; Wang, H. DNA methylome analysis provides evidence that the expansion of the tea genome is linked to TE bursts. Plant Biotechnol. J. 2019, 17, 826–835. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, H.; Khemka, N.; Jain, M.; Garg, R. Genome-wide bisulphite-sequencing reveals organ-specific methylation patterns in chickpea. Sci. Rep. 2018, 8, 9704. [Google Scholar] [CrossRef] [PubMed]
- An, Y.C.; Goettel, W.; Han, Q.; Bartels, A.; Liu, Z.; Xiao, W. Dynamic Changes of Genome-Wide DNA Methylation during Soybean Seed Development. Sci. Rep. 2017, 7, 12263. [Google Scholar] [CrossRef]
- Wang, L.; Xie, J.; Hu, J.; Lan, B.; You, C.; Li, F.; Wang, Z.; Wang, H. Comparative epigenomics reveals evolution of duplicated genes in potato and tomato. Plant J. 2018, 93, 460–471. [Google Scholar] [CrossRef] [Green Version]
- Hauser, M.T.; Aufsatz, W.; Jonak, C.; Luschnig, C. Transgenerational epigenetic inheritance in plants. Biochim. Biophys. Acta 2011, 1809, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Zemach, A.; Zilberman, D. Evolution of eukaryotic DNA methylation and the pursuit of safer sex. Curr. Biol. 2010, 20, R780–R785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmeister, B.T.; Lee, K.; Rohr, N.A.; Hall, D.W.; Schmitz, R.J. Stable inheritance of DNA methylation allows creation of epigenotype maps and the study of epiallele inheritance patterns in the absence of genetic variation. Genome Biol. 2017, 18, 155. [Google Scholar] [CrossRef]
- Wang, G.; Köhler, C. Epigenetic processes in flowering plant reproduction. J. Exp. Bot. 2017, 68, 797–807. [Google Scholar] [CrossRef] [Green Version]
- Chow, H.T.; Chakraborty, T.; Mosher, R.A. RNA-directed DNA Methylation and sexual reproduction: Expanding beyond the seed. Curr. Opin. Plant Biol. 2020, 54, 11–17. [Google Scholar] [CrossRef]
- Jacobsen, S.E.; Meyerowitz, E.M. Hypermethylated superman epigenetic alleles in Arabidopsis. Science 1997, 277, 1100–1103. [Google Scholar] [CrossRef] [Green Version]
- Soppe, W.J.J.; Jacobsen, S.E.; Alonso-Blanco, C.; Jackson, J.P.; Kakutani, T.; Koornneef, M.; Peeters, A.J.M. The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol. Cell 2000, 6, 791–802. [Google Scholar] [CrossRef]
- Stokes, T.L.; Kunkel, B.N.; Richards, E.J. Epigenetic variation in Arabidopsis disease resistance. Genes Dev. 2002, 16, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Cocciolone, S.M.; Chopra, S.; Flint-Garcia, S.A.; McMullen, M.D.; Peterson, T. Tissue-specific patterns of a maize Myb transcription factor are epigenetically regulated. Plant J. 2001, 27, 467–478. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Wu, W.; Zinta, G.; Yang, L.; Wang, D.; Liu, R.; Zhang, H.; Zheng, Z.; Huang, H.; Zhang, Q.; et al. A naturally occurring epiallele associates with leaf senescence and local climate adaptation in Arabidopsis accessions. Nat. Commun. 2018, 9, 460. [Google Scholar] [CrossRef] [PubMed]
- Johannes, F.; Porcher, E.; Teixeira, F.K.; Saliba-Colombani, V.; Simon, M.; Agier, N.; Bulski, A.; Albuisson, J.; Heredia, F.; Audigier, P.; et al. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 2009, 5, e1000530. [Google Scholar] [CrossRef]
- Cortijo, S.; Wardenaar, R.; Colome-Tatche, M.; Gilly, A.; Etcheverry, M.; Labadie, K.; Caillieux, E.; Hospital, F.; Aury, J.M.; Wincker, P.; et al. Mapping the epigenetic basis of complex traits. Science 2014, 343, 1145–1148. [Google Scholar] [CrossRef]
- Slatkin, M. Epigenetic inheritance and the missing heritability problem. Genetics 2009, 182, 845–850. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.; Weigel, D. Epigenetic variation: Origin and transgenerational inheritance. Curr. Opin. Plant Biol. 2012, 15, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Cubas, P.; Vincent, C.; Coen, E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature 1999, 401, 157–161. [Google Scholar] [CrossRef]
- Manning, K.; Tor, M.; Poole, M.; Hong, Y.; Thompson, A.J.; King, G.J.; Giovannoni, J.J.; Seymour, G.B. A naturally occurring epigenetic mutation in a gene encoding an sbp-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 2006, 38, 948–952. [Google Scholar] [CrossRef]
- Quadrana, L.; Almeida, J.; Asís, R.; Duffy, T.; Dominguez, P.G.; Bermúdez, L.; Conti, G.; Corrêa da Silva, J.V.; Peralta, I.E.; Colot, V.; et al. Natural occurring epialleles determine vitamin E accumulation in tomato fruits. Nat. Commun. 2014, 5, 3027. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.; Troadec, C.; Boualem, A.; Rajab, M.; Fernandez, R.; Morin, H.; Pitrat, M.; Dogimont, C.; Bendahmane, A. A transposon-induced epigenetic change leads to sex determination in melon. Nature 2009, 461, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Shiba, H.; Kakizaki, T.; Iwano, M.; Tarutani, Y.; Watanabe, M.; Isogai, A.; Takayama, S. Dominance relationships between self-incompatibility alleles controlled by DNA methylation. Nat. Genet. 2006, 38, 297–299. [Google Scholar] [CrossRef]
- Bender, J.; Fink, G.R. Epigenetic control of an endogenous gene family is revealed by a novel blue fluorescent mutant of Arabidopsis. Cell 1995, 83, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, S.E.; Sakai, H.; Finnegan, E.J.; Cao, X.; Meyerowitz, E.M. Ectopic hypermethylation of flower specific genes in Arabidopsis. Curr. Biol. 2000, 24, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Saze, H.; Shiraishi, A.; Miura, A.; Kakutani, T. Control of genic DNA methylation by a jmjc domain-containing protein in Arabidopsis thaliana. Science 2008, 319, 462–465. [Google Scholar] [CrossRef]
- Durand, S.; Bouche, N.; Perez, S.E.; Loudet, O.; Camilleri, C. Rapid establishment of genetic incompatibility through natural epigenetic variation. Curr. Biol. 2012, 22, 326–331. [Google Scholar] [CrossRef] [Green Version]
- Silveira, A.B.; Trontin, C.; Cortijo, S.; Barau, J.; Del Bem, L.E.; Loudet, O.; Colot, V.; Vincentz, M. Extensive natural epigenetic variation at a de novo originated gene. PLoS Genet. 2013, 9, e1003437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blevins, T.; Wang, J.; Pflieger, D.; Pontvianne, F.; Pikaard, C.S. Hybrid incompatibility caused by an epiallele. Proc. Natl. Acad. Sci. USA 2017, 114, 3702–3707. [Google Scholar] [CrossRef] [Green Version]
- Brink, R.A. A Genetic Change Associated with the R Locus in Maize Which Is Directed and Potentially Reversible. Genetics 1956, 41, 872–889. [Google Scholar]
- Patterson, G.I.; Thorpe, C.J.; Chandler, V.L. Paramutation, an allelic interaction, is associated with a stable and heritable reduction of transcription of the maize b regulatory gene. Genetics 1993, 135, 881–894. [Google Scholar]
- Hollick, J.B.; Patterson, G.I.; Coe, E.H.; Cone, K.C.; Chandler, V.L. Allelic interactions heritably influence the activity of a metastable maize pl allele. Genetics 1995, 141, 709–719. [Google Scholar] [PubMed]
- Pilu, R.; Panzeri, D.; Cassani, E.; Cerino Badone, F.; Landoni, M.; Nielsen, E.A. paramutation phenomenon is involved in the genetics of maize low phytic acid1-241 (lpa1-241) trait. Heredity 2009, 102, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Agetsuma, M.; Kitano, H.; Yoshimura, A.; Matsuoka, M.; Jacobsen, S.E.; Ashikari, M. A metastable dwarf1 epigenetic mutant affecting plant stature in rice. Proc. Natl. Acad. Sci. USA 2009, 106, 11218–11223. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Ikeda, M.; Matsubara, A.; Song, X.J.; Ito, M.; Asano, K.; Matsuoka, M.; Kitano, H.; Ashikari, M. Osspl14 promotes panicle branching and higher grain productivity in rice. Nat. Genet. 2010, 42, 545–549. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, Z.; Qin, R.; Qiu, Y.; Wang, J.L.; Cui, X.; Gu, L.; Zhang, X.; Guo, X.; Wang, D.; et al. Identification and characterization of an epi-allele of fie1 reveals a regulatory linkage between two epigenetic marks in rice. Plant Cell 2012, 24, 4407–4421. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Sun, J.; Cao, X.; Song, X. Epigenetic Mutation of RAV6 Affects Leaf Angle and Seed Size in Rice. Plant Physiol. 2015, 169, 2118–2128. [Google Scholar]
- Wei, X.; Song, X.; Wei, L.; Tang, S.; Sun, J.; Hu, P.; Cao, X. An epiallele of rice AK1 affects photosynthetic capacity. J. Integr. Plant Biol. 2017, 59, 158–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, X.; Liu, S.; Ke, S.; Dai, H.; Xie, X.M.; Hsieh, T.F.; Zhang, X.Q. Epigenetic modification of ESP, encoding a putative long noncoding RNA, affects panicle architecture in rice. Rice 2019, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Ong-Abdullah, M.; Ordway, J.M.; Jiang, N.; Ooi, S.E.; Kok, S.Y.; Sarpan, N.; Azimi, N.; Hashim, A.T.; Ishak, Z.; Rosli, S.K.; et al. Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature 2015, 525, 533–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wendte, J.M.; Ji, L.; Schmitz, R.J. Natural variation in DNA methylation homeostasis and the emergence of epialleles. Proc. Natl. Acad. Sci. USA 2020, 117, 4874–4884. [Google Scholar] [CrossRef]
- Mirouze, M.; Paszkowski, J. Epigenetic contribution to stress adaptation in plants. Curr. Opin. Plant Biol. 2011, 14, 267–274. [Google Scholar] [CrossRef]
- Kooke, R.; Keurentjes, J.J.B. Epigenetic variation contributes to environmental adaptation of Arabidopsis thaliana. Plant Signal. Behav. 2015, 10, e1057368. [Google Scholar] [CrossRef] [Green Version]
- Reinders, J.; Wulff, B.B.; Mirouze, M.; Marí-Ordonez, A.; Dapp, M.; Rozhon, W.; Bucher, E.; Theiler, G.; Paszkowski, J. Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev. 2009, 23, 939–950. [Google Scholar] [CrossRef] [Green Version]
- Kankel, M.W.; Ramsey, D.E.; Stokes, T.L.; Flowers, S.K.; Haag, J.R.; Jeddeloh, J.A.; Riddle, N.C.; Verbsky, M.L.; Richards, E.J. Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 2003, 163, 1109–1122. [Google Scholar]
- Jeddeloh, J.A.; Stokes, T.L.; Richards, E.J. Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat Genet. 1999, 22, 94–97. [Google Scholar] [CrossRef]
- Lippman, Z.; Gendrel, A.V.; Black, M.; Vaughn, M.W.; Dedhia, N.; McCombie, W.R.; Lavine, K.; Mittal, V.; May, B.; Kasschau, K.D.; et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004, 430, 471–476. [Google Scholar] [CrossRef]
- Vongs, A.; Kakutani, T.; Martienssen, R.A.; Richards, E.J. Arabidopsis thaliana DNA methylation mutants. Science 1993, 260, 1926–1928. [Google Scholar] [CrossRef]
- Kakutani, T.; Munakata, K.; Richards, E.J.; Hirochika, H. Meiotically and mitotically stable inheritance of DNA hypomethylation induced by ddm1 mutation of Arabidopsis thaliana. Genetics 1999, 151, 831–838. [Google Scholar]
- Teixeira, F.K.; Heredia, F.; Sarazin, A.; Roudier, F.; Boccara, M.; Ciaudo, C.; Cruaud, C.; Poulain, J.; Berdasco, M.; Fraga, M.F.; et al. A role for RNAi in the selective correction of DNA methylation defects. Science 2009, 323, 1600–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Yang, D.L.; Huang, H.; Zhang, G.; He, L.; Pang, J.; Lozano-Durán, R.; Lang, Z.; Zhu, J.K. Epigenetic memory marks determine epiallele stability at loci targeted by de novo DNA methylation. Nat. Plants 2020, 6, 661–674. [Google Scholar] [CrossRef]
- McKeown, P.C.; Fort, A.; Duszynska, D.; Sulpice, R.; Spillane, C. Emerging molecular mechanisms for biotechnological harnessing of heterosis in crops. Trends Biotechnol. 2013, 31, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Genomic and epigenetic insights into the molecular bases of heterosis. Nat. Rev. Genet. 2013, 14, 471–482. [Google Scholar] [CrossRef]
- Groszmann, M.; Greaves, I.K.; Fujimoto, R.; Peacock, W.J.; Dennis, E.S. The role of epigenetics in hybrid vigour. Trends Genet. 2013, 29, 684–690. [Google Scholar] [CrossRef]
- Lauss, K.; Wardenaar, R.; Oka, R.; van Hulten, M.; Guryev, V.; Keurentjes, J.; Stam, M.; Johannes, F. Parental DNA Methylation States are associated with Heterosis in Epigenetic Hybrids. Plant Physiol. 2018, 176, 1627–1645. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; He, H.; Li, J.; Chen, W.; Wang, X.; Guo, L.; Peng, Z.; He, G.; Zhong, S.; Qi, Y.; et al. Genome-wide analysis of DNA methylation and gene expression changes in two Arabidopsis ecotypes and their reciprocal hybrids. Plant Cell 2012, 24, 875–892. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, D.; Lang, Z.; He, L.; Yang, L.; Zeng, L.; Li, Y.; Zhao, C. Methylation interactions in Arabidopsis hybrids require RNA-directed DNA methylation and are influenced by genetic variation. Proc. Natl. Acad. Sci. USA 2016, 113, E4248–E4256. [Google Scholar] [CrossRef] [Green Version]
- Groszmann, M.; Greaves, I.K.; Albertyn, Z.I.; Scofield, G.N.; Peacock, W.J.; Dennis, E.S. Changes in 24-nt siRNA levels in Arabidopsis hybrids suggest an epigenetic contribution to hybrid vigor. Proc. Natl. Acad. Sci. USA 2011, 108, 2617–2622. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, B.; Duan, C.G.; Zhu, J.K. Chemical probes in plant epigenetics studies. Plant Signal Behav. 2013, 8, e25364. [Google Scholar] [CrossRef] [Green Version]
- Amoah, S.; Kurup, S.; Lopez, C.M.R.; Welham, S.J.; Powers, S.J.; Hopkins, C.J.; Wilkinson, M.J.; King, G.J. A Hypomethylated population of Brassica rapa for forward and reverse Epi-genetics. BMC Plant Biol. 2012, 12, 193. [Google Scholar] [CrossRef] [Green Version]
- Tal, O.; Kisdi, E.; Jablonka, E. Epigenetic contribution to covariance between relatives. Genetics 2010, 184, 1037–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannes, F.; Colome-Tatche, M. Quantitative epigenetics through epigenomic perturbation of isogenic lines. Genetics 2011, 188, 215–227. [Google Scholar] [CrossRef] [Green Version]
- Furrow, R.E.; Christiansen, F.B.; Feldman, M.W. Environment-sensitive epigenetics and the heritability of complex diseases. Genetics 2011, 189, 1377–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, E.; Goggins, M. DNA methylation analysis in human cancer. Methods Mol. Biol. 2013, 980, 131–156. [Google Scholar]
- Estival, A.; Sanz, C.; Ramirez, J.L.; Velarde, J.M.; Domenech, M.; Carrato, C.; de Las Peñas, R.; Gil-Gil, M.; Sepúlveda, J.; Armengol, R.; et al. Pyrosequencing versus methylation-specific PCR for assessment of MGMT methylation in tumor and blood samples of glioblastoma patients. Sci. Rep. 2019, 9, 11125. [Google Scholar] [CrossRef]
- Munson, K.; Clark, J.; Lamparska-Kupsik, K.; Smith, S.S. Recovery of bisulfite-converted genomic sequences in the methylation-sensitive QPCR. Nucleic Acids Res. 2007, 35, 2893–2903. [Google Scholar] [CrossRef] [Green Version]
- Hernández, H.G.; Tse, M.Y.; Pang, S.C.; Arboleda, H.; Forero, D.A. Optimizing methodologies for PCR-based DNA methylationn analysis. Biotechniques 2013, 55, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Kanherkar, R.R.; Bhatia-Dey, N.; Csoka, A.B. Epigenetics across the human lifespan. Front. Cell Dev. Biol. 2014, 2, 49. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, A.E.; Feinberg, A.P.; Irizarry, R.A.; Leek, J.T. Significance analysis and statistical dissection of variably methylated regions. Biostatistics 2012, 13, 166–178. [Google Scholar] [CrossRef] [Green Version]
- Roux, F.; Colome-Tatche, M.; Edelist, C.; Warenaar, R.; Guerche, P.; Hospital, F.; Colot, V.; Jansen, R.C.; Johannes, F. Genome-wide epigenetic perturbation jump-starts patterns of heritable variation found in nature. Genetics 2011, 188, 1015–1017. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, Z.; Wang, J.; Sui, Y.; Zhang, J.; Liao, D.; Wu, R. A quantitative genetic and epigenetic model of complex traits. BMC Bioinform. 2012, 13, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, Y.; Xia, W.; Li, R.; Wang, J.; Shao, M.; Feng, J.; King, G.J.; Meng, J. Epigenetic QTL mapping in Brassica napus. Genetics 2011, 189, 1093–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Y.; Qian, J.; Sun, Y.; Yi, Z.; Yan, M. Construction of methylation linkage map based on MSAP and SSR markers in Sorghum bicolor (L.). IUBMB Life 2009, 61, 663–669. [Google Scholar] [CrossRef]
- Schmitz, R.J.; He, Y.; Lopez, O.V.; Khan, S.M.; Joshi, T.; Urich, M.A.; Nery, J.R.; Diers, B.; Xu, D.; Stacey, G.; et al. Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 2013, 23, 1663–1674. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; McKnight, A.J.; Craig, D.; O’Neill, F. Epigenome-Wide Association Study for Parkinson’s Disease. Neuro Mol. Med. 2014, 16, 845–855. [Google Scholar] [CrossRef]
- Sharma, P.; Garg, G.; Kumar, A.; Mohammad, F.; Kumar, S.R.; Tanwar, V.S.; Sati, S.; Sharma, A.; Karthikeyan, G.; Brahmachari, V.; et al. Genome wide DNA methylation profiling for epigenetic alteration in coronary artery disease patients. Gene 2014, 541, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Gasparoni, G.; Bultmann, S.; Lutsik, P.; Kraus, T.; Sordon, S.; Vlcek, J.; Dietinger, V.; Steinmaurer, M.; Haider, M.; Mulholland, C.B.; et al. DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenetics Chromatin 2018, 11, 41. [Google Scholar] [CrossRef]
- Cardona, A.; Day, F.R.; Perry, J.; Loh, M.; Chu, A.Y.; Lehne, B.; Paul, D.S.; Lotta, L.A.; Stewart, I.D.; Kerrison, N.D.; et al. Epigenome-Wide Association Study of Incident Type 2 Diabetes in a British Population: EPIC-Norfolk Study. Diabetes 2019, 68, 2315–2326. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zou, D.; Li, Z.; Gao, R.; Sang, J.; Zhang, Y.; Li, R.; Xia, L.; Zhang, T.; Niu, G.; et al. EWAS Atlas: A curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res. 2019, 47, D983–D988. [Google Scholar] [CrossRef]
- Lu, W.; Xiao, L.; Quan, M.; Wang, Q.; El-Kassaby, Y.A.; Du, Q.; Zhang, D. Linkage-linkage disequilibrium dissection of the epigenetic quantitative trait loci (epiQTLs) underlying growth and wood properties in Populus. New Phytol. 2020, 225, 1218–1233. [Google Scholar] [CrossRef]
- Schield, D.R.; Walsh, M.R.; Card, D.C.; Andrew, A.L.; Adams, R.H.; Castoe, T.A. EpiRADseq: Scalable analysis of genomewide patterns of methylation using next-generation sequencing. Methods Ecol. Evol. 2016, 7, 60–69. [Google Scholar] [CrossRef]
- Trucchi, E.; Mazzarella, A.B.; Gilfillan, G.D.; Lorenzo, M.T.; Schönswetter, P.; Paun, O. BsRADseq: Screening DNA methylation in natural populations of non-model species. Mol. Ecol. 2016, 25, 1697–1713. [Google Scholar] [CrossRef] [Green Version]
- van Gurp, T.P.; Wagemaker, N.C.; Wouters, B.; Vergeer, P.; Ouborg, J.N.; Verhoeven, K.J. epiGBS: Reference-free reduced representation bisulfite sequencing. Nat. Methods 2016, 13, 322–324. [Google Scholar] [CrossRef]
- Werner, O.; Prudencio, Á.S.; de la Cruz-Martínez, E.; Nieto-Lugilde, M.; Martínez-Gómez, P.; Ros, R.M. A Cost Reduced Variant of Epi-Genotyping by Sequencing for Studying DNA Methylation in Non-model Organisms. Front. Plant Sci. 2020, 11, 694. [Google Scholar] [CrossRef]
- Prudencio, Á.S.; Werner, O.; Martínez-García, P.J.; Dicenta, F.; Ros, R.M.; Martínez-Gómez, P. DNA Methylation Analysis of Dormancy Release in Almond (Prunus dulcis) Flower Buds Using Epi-Genotyping by Sequencing. Int. J. Mol. Sci. 2018, 19, 3542. [Google Scholar] [CrossRef] [Green Version]
- Gallego-Bartolomé, J. DNA methylation in plants: Mechanisms and tools for targeted manipulation. New Phytol. 2020, 227, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.M.; Du, J.; Hale, C.J.; Bischof, S.; Feng, S.; Chodavarapu, R.K.; Zhong, X.; Marson, G.; Pellegrini, M.; Segal, D.J.; et al. SRA- and SET-domain-containing proteins link RNA polymerase V occupancy to DNA methylation. Nature 2014, 507, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Gallego-Bartolomé, J.; Liu, W.; Kuo, P.H.; Feng, S.; Ghoshal, B.; Gardiner, J.; Zhao, J.M.; Park, S.Y.; Chory, J.; Jacobsen, S.E. Co-targeting RNA Polymerases IV and V Promotes Efficient De Novo DNA Methylation in Arabidopsis. Cell 2019, 176, 1068–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papikian, A.; Liu, W.; Gallego-Bartolomé, J.; Jacobsen, S.E. Site-specific manipulation of Arabidopsis loci using CRISPR-Cas9 SunTag systems. Nat. Commun. 2019, 10, 729. [Google Scholar] [CrossRef]
- Gallego-Bartolomé, J.; Gardiner, J.; Liu, W.; Papikian, A.; Ghoshal, B.; Kuo, H.Y.; Zhao, J.M.; Segal, D.J.; Jacobsen, S.E. Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain. Proc. Natl. Acad. Sci. USA 2018, 115, E2125–E2134. [Google Scholar] [CrossRef] [Green Version]
- Shew, A.M.; Nallwy, L.L.; Snell, H.A.; Nayga, R.M.; Dixon, B.L. CRISPR Versus GMOs: Public Acceptance and Valuation. Glob. Food Secur. 2018, 19, 71–80. [Google Scholar] [CrossRef]
- Ishii, T.; Araki, M. Consumer acceptance of food crops developed by genome editing. Plant Cell Rep. 2016, 35, 1507–1518. [Google Scholar] [CrossRef]
- Amabile, A.; Migliara, A.; Capasso, P.; Biffi, M.; Cittaro, D.; Naldini, L.; Lombardo, A. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016, 167, 219–232.e14. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247.e17. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Species (Common Name) | Family | Monocot/Eudicot | Genome Size (Mb) | Repeat Elements | CpG (%) | CpHpG (%) | CpHpH (%) | Reference |
|---|---|---|---|---|---|---|---|---|
| Arabidopsis thaliana (Arabidopsis) | Brassicaceae | Eudicot | 135 | 31,189 | 24.00 | 6.70 | 1.70 | [15] |
| Beta vulgaris (beet) | Amaranthaaceae | Eudicot | 758 | 656,014 | 92.00 | 81.00 | 18.80 | [4] |
| Brassica oleracea (cabbage) | Brassicaceae | Eudicot | 648 | 532,987 | 52.50 | 22.00 | 5.11 | [4] |
| Brassica rapa (mustard) | Brassicaceae | Eudicot | 485 | 218,781 | 37.20 | 17.28 | 4.44 | [4] |
| Cajanus cajan (pigeonpea) | Fabaceae | Eudicot | 833 | 1,127,729 | 70.23 | 54.60 | 9.87 | [19] |
| Camellia sinensis (tea) | Theaceae | Eudicot | 3100 | 5,164,785 | 82.00 | 70.00 | 10.00 | [20] |
| Cannabis sativa (canabis) | Cannabaceae | Eudicot | 818 | 376,401 | 75.50 | 65.00 | 8.72 | [4] |
| Capsella rubella (pink shepherd’s purse) | Brassicaceae | Eudicot | 219 | 39,716 | 32.00 | 9.90 | 34.70 | [4] |
| Cicer arietinum (chickpea) | Fabaceae | Eudicot | 738 | 853,514 | 93.00 | 89.00 | 38.00 | [21] |
| Citrus clementina (clementine) | Rutaceae | Eudicot | 370 | 205,699 | 45.83 | 25.13 | 8.26 | [4] |
| Cucumis sativus (cucumber) | Cucurbitaceae | Eudicot | 367 | 57,750 | 45.88 | 16.50 | 4.12 | [4] |
| Eucalyptus grandis (rose gum) | Myrtaceae | Eudicot | 640 | 689,306 | 37.12 | 19.96 | 1.36 | [4] |
| Fragaria vesca (strawberry) | Rosaceae | Eudicot | 240 | 129,500 | 48.35 | 20.63 | 2.32 | [4] |
| Glycine max (soybean) | Fabaceae | Eudicot | 1115 | 38,581 | 63.20 | 38.40 | 4.10 | [22] |
| Gossypium raimondii (cotton) | Malvaceae | Eudicot | 880 | 489,564 | 71.97 | 57.80 | 13.14 | [4] |
| Lotus japonicus (birdsfoot trefoil) | Fabaceae | Eudicot | 472 | 160,505 | 67.75 | 36.59 | 8.66 | [4] |
| Malus domestica (apple) | Rosaceae | Eudicot | 742 | 1,245,768 | 63.50 | 44.14 | 4.57 | [4] |
| Manihot esculenta (cassava) | Euphorbiaceae | Eudicot | 742 | 258,416 | 51.53 | 30.38 | 1.90 | [4] |
| Medicago truncatula (barrel clover) | Fabaceae | Eudicot | 465 | 375,003 | 59.80 | 16.94 | 5.09 | [4] |
| Populus trichocarpa (poplar) | Salicaceae | Eudicot | 500 | 173,230 | 43.95 | 26.78 | 5.01 | [4] |
| Prunus persica (peach) | Rosaceae | Eudicot | 265 | 95,678 | 50.18 | 19.59 | 3.64 | [4] |
| Ricinus communis (castor bean) | Euphorbiaceae | Eudicot | 323 | 575,449 | 64.54 | 37.94 | 11.97 | [4] |
| Solanaceae lycopersicum (tomato) | Solanaceae | Eudicot | 907 | 887,009 | 84.05 | 54.84 | 8.35 | [4] |
| Solanaceae tuberosum (potato) | Solanaceae | Eudicot | 840 | 404,861 | 70.90 | 42.20 | 15.80 | [23] |
| Vitis vinifera (grape vine) | Vitaceae | Eudicot | 487 | 449,466 | 45.95 | 20.43 | 1.15 | [4] |
| Brachypodium distachyon (stiff brome) | Poaceae | Monocot | 352 | 51,793 | 49.17 | 19.17 | 1.41 | [4] |
| Oryza sativa (rice) | Poaceae | Monocot | 430 | 447,163 | 54.70 | 37.30 | 12.00 | [16] |
| Panicum hallii (Hall’s panicgrass) | Poaceae | Monocot | 550 | 154,970 | 56.28 | 29.97 | 2.43 | [4] |
| Panicum virgatum (switchgrass) | Poaceae | Monocot | 1600 | 1,793,620 | 53.56 | 35.74 | 3.06 | [4] |
| Setaria viridis (green foxtail) | Poaceae | Monocot | 515 | 372,068 | 44.49 | 23.25 | 1.56 | [4] |
| Sorghum bicolor (sorghum) | Poaceae | Monocot | 730 | 397,003 | 84.75 | 73.25 | 5.81 | [4] |
| Triticum aestivum (wheat) | Poaceae | Monocot | 17,000 | 3,968,974 | 53.30 | 3.48 | 1.41 | [17] |
| Zea mays (maize) | Poaceae | Monocot | 2665 | 1,971,471 | 86.00 | 74.00 | 5.40 | [7] |
| Species | Gene/Locus | Epigenetic Variation | Phenotypic Traits | References |
|---|---|---|---|---|
| Arabidopsis thaliana | SUP (SUPERMAN) | Mutagen induced | Increased numbers of stamens and carpels | [29] |
| FWA (Flowering Wageningen) | Mutagen induced | Late flowering | [30] | |
| PAI2 (Phosphoribosyl Anthranilate Isomerise) | Trans-acting (small RNAs) | Only gene expression affected; no specific phenotype | [43] | |
| BAL1 | Mutagen induced | Dwarfing and elevated disease resistance | [31] | |
| AG (AGAMOUS) | Mutagen induced | Affect flower structure | [44] | |
| BNS (BONSAI) | ddm1-induced syndrome | Stunted growth | [45] | |
| FOLT1(folate transporter 1) | Trans-acting (small RNAs) | Reduced fertility | [46] | |
| QQS (Qua-Quine Starch) | Spontaneous | Higher starch accumulation | [47] | |
| PPH (Pheophytin Pheophorbide Hydrolase) | Spontaneous | Inhibits leaf senescence | [33] | |
| HISN6B (Histidinol-phosphate aminotransferase 1) | Spontaneous | Hybrid incompatibility | [48] | |
| Zea Mays | r1 (red1) | Spontaneous | Reduced pigmentation | [49] |
| b1(booster 1) | Spontaneous | Reduced pigmentation | [50] | |
| pl1 (purple plant 1) | Spontaneous | Reduced pigmentation | [51] | |
| p1 (pericarp color 1) | Spontaneous | Reduced pigmentation | [32] | |
| lpa1(low phytic acid1) | Paramutagenic | High inorganic phosphate in seeds | [52] | |
| Linaria vulgaris | Lcyc (Linaria cycliodea) | Spontaneous | Floral symmetry; dorsiventral flower axis | [38] |
| Solanum lycopersicum | CNR (Colorless non-ripening) | Spontaneous | Normal fruit ripening | [39] |
| VTE3 (Vitamin E) | Spontaneous | Tocopherol accumulation in fruit | [40] | |
| Oryza sativa | D1 (Drawf1) | Spontaneous | Dwarf | [53] |
| SPL14 (Squamosa Promoter binding protein-Like) | Spontaneous | Panicle branching and higher grain yield | [54] | |
| FIE1 (Fertilization-Independent Endosperm 1) | Spontaneous | Dwarf | [55] | |
| RAV6 [Related to Abscisic Acid Insensitive 3 (ABI3)/Viviparous1 (VP1) 6] | Spontaneous | Larger lamina inclination and smaller grain size | [56] | |
| AK1 (Adenylate Kinase 1) | Spontaneous | Defects in photosynthetic capacity | [57] | |
| ESP (Epigenetic Short Panicle) | Spontaneous | Short panicle | [58] | |
| Elaeis guineensis | DEF1 (DEFICIENS) | Spontaneous | Mantled fruit | [59] |
| Brassica rapa | SP11/SCR (S locus protein 11/S locus cystein rich) | Trans-acting (small RNAs) | Self-incompatibility | [42] |
| Cucumis melo | CmWIP1 (WASP/N-WASP-interacting protein 1) | Transposon Insertion | Sex determination | [41] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gahlaut, V.; Zinta, G.; Jaiswal, V.; Kumar, S. Quantitative Epigenetics: A New Avenue for Crop Improvement. Epigenomes 2020, 4, 25. https://doi.org/10.3390/epigenomes4040025
Gahlaut V, Zinta G, Jaiswal V, Kumar S. Quantitative Epigenetics: A New Avenue for Crop Improvement. Epigenomes. 2020; 4(4):25. https://doi.org/10.3390/epigenomes4040025
Chicago/Turabian StyleGahlaut, Vijay, Gaurav Zinta, Vandana Jaiswal, and Sanjay Kumar. 2020. "Quantitative Epigenetics: A New Avenue for Crop Improvement" Epigenomes 4, no. 4: 25. https://doi.org/10.3390/epigenomes4040025