
Genome-Wide Epigenetic Studies in Chicken: A Review

 and
and 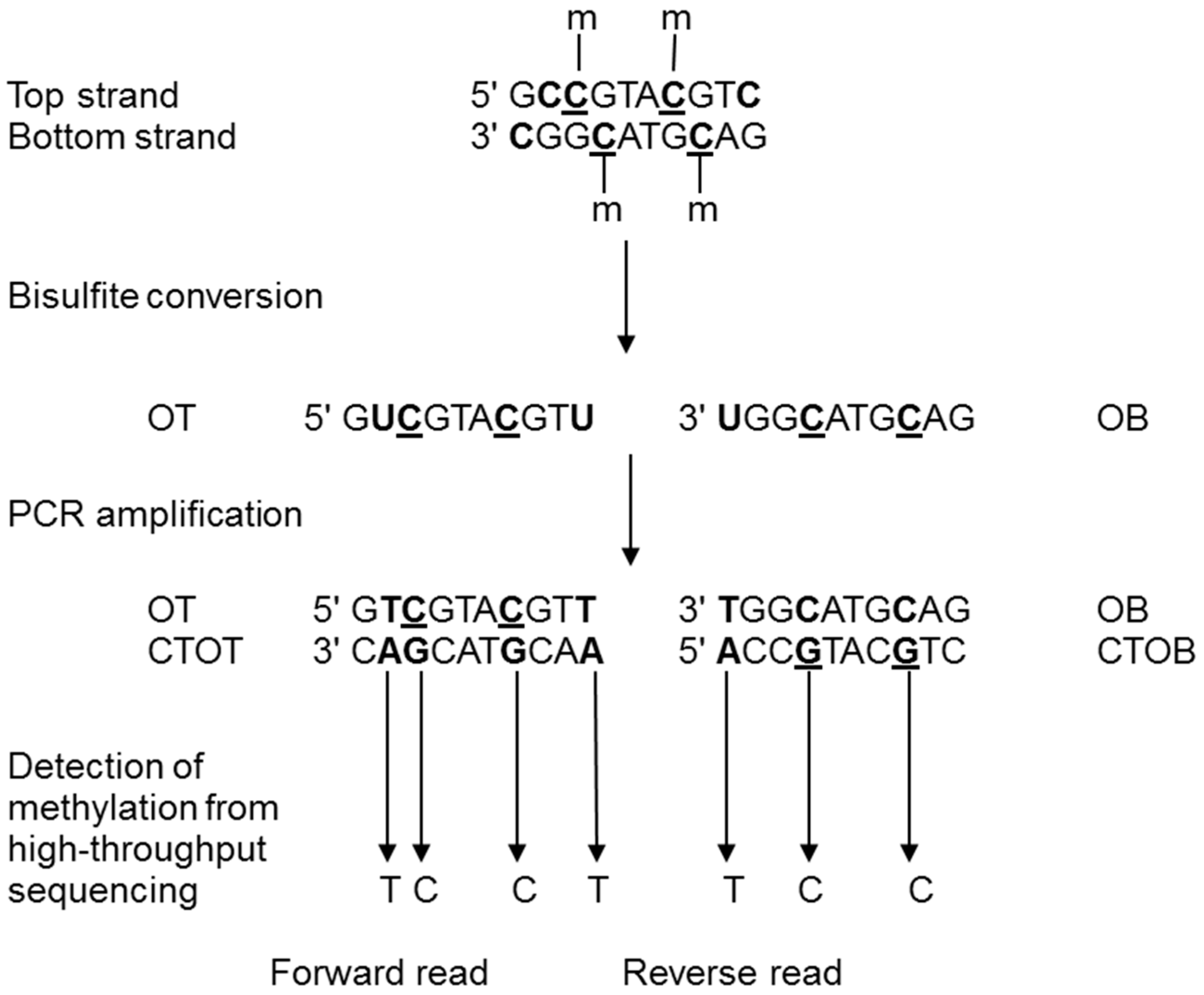{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. From Epigenetics to Epigenomics
2.1. Epigenetic Marks
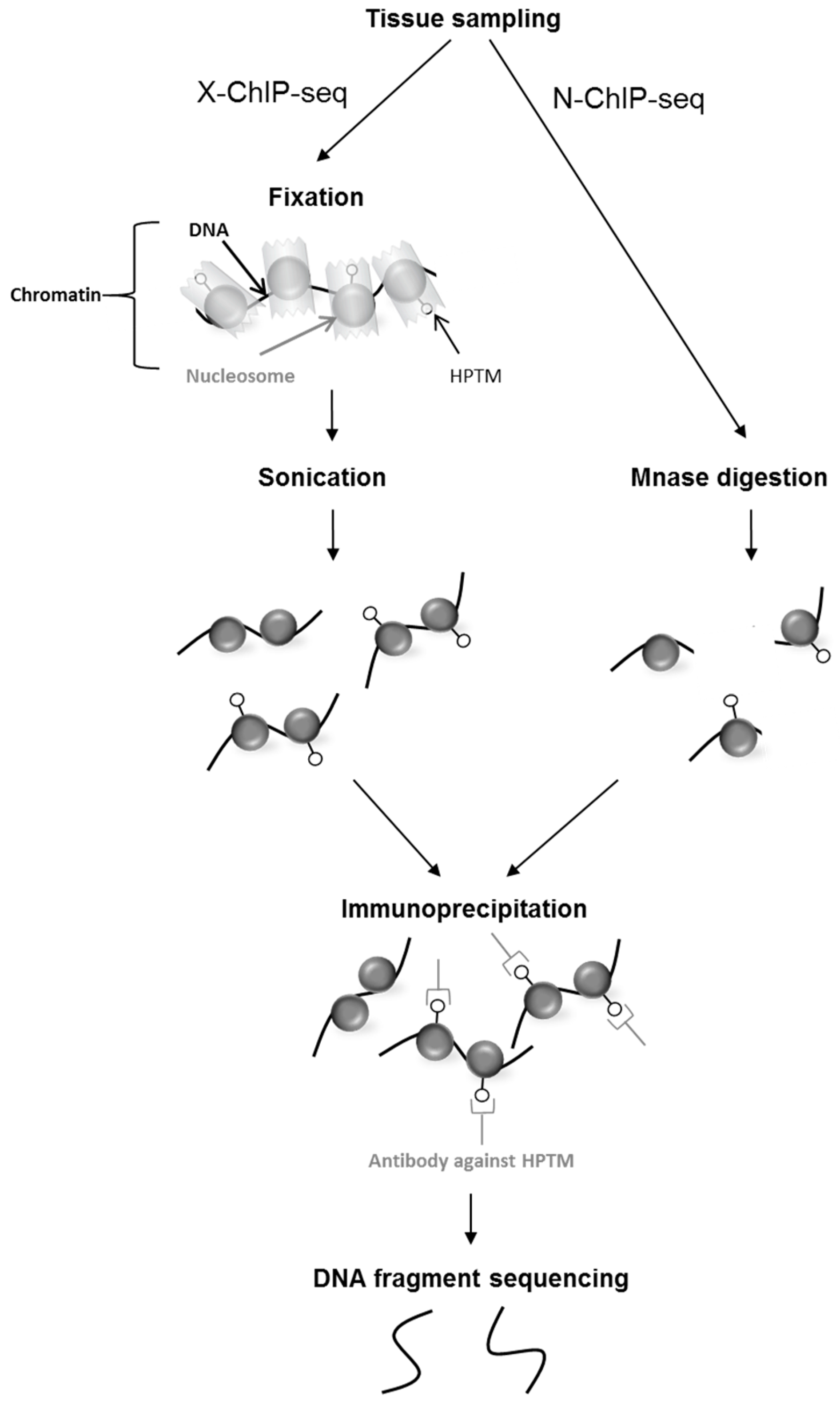
2.2. Epigenomics Methods
3. New Knowledge Gained on Chicken Models by High-Throughput Sequencing
3.1. The Chicken Genome
3.2. The chicken DNA Methylation Landscape
3.3. The Chicken HPTM Landscape
4. Design of Epigenomic Studies
4.1. Sequencing Considerations
4.2. Bioinformatic Data Analysis
5. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 5hmC | 5-hydroxymethylcytosine |
| 5mC | 5-methylcytosine |
| BS-seq | Bisulfite Sequencing |
| ChIP-seq | Chromatin ImmunoPrecipitation followed by sequencing |
| ENCODE | ENCyclopedia of DNA Elements |
| FAANG | Functional Annotation of ANimal Genomes |
| Go | Giga-octet |
| H3K27ac | histone H3 lysine 27 acetylation |
| H3K27me3 | histone H3 lysine 27 tri-methylation |
| H3k36me3 | histone H3 lysine 36 tri-methylation |
| H3K4me3 | histone H3 lysine 4 tri-methylation |
| H3K9me3 | histone H3 lysine 9 tri-methylation |
| HPTM | Histone Post Translational Modifications |
| HTS | High-Throughput Sequencing |
| IHEC | International Human Epigenome Consortium |
| Ko | Kilo-octet |
| MBD-seq | Methyl Binding Domain sequencing |
| MeDIP-seq | MEthylated DNA ImmunoPrecipitation sequencing |
| MethylC-seq | MethylC-sequencing |
| modENCODE | Model Organism ENCyclopedia Of DNA Elements |
| MRE-seq | Methyl-sensitive Restriction Enzyme digestion sequencing |
| N-ChIP-seq | Native Chromatin ImmunoPrecipitation followed by sequencing |
| RRBS | Reduced Representation Bisulfite Sequencing |
| TSS | Transcription Start Site |
| UTR | Untranslated Transcribed Region |
| WGBS | Whole Genome Bisulfite Sequencing |
| X-ChIP-seq | Cross-linked Chromatin ImmunoPrecipitation followed by sequencing |
References
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Triantaphyllopoulos, K.A.; Ikonomopoulos, I.; Bannister, A.J. Epigenetics and inheritance of phenotype variation in livestock. Epigenet. Chromatin 2016, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Nätt, D.; Lindqvist, N.; Stranneheim, H.; Lundeberg, J.; Torjesen, P.A.; Jensen, P. Inheritance of acquired behaviour adaptations and brain gene expression in chickens. PLoS ONE 2009, 4, e6405. [Google Scholar] [CrossRef]
- Verhulst, E.C.; Mateman, A.C.; Zwier, M.V.; Caro, S.P.; Verhoeven, K.J.F.; van Oers, K. Evidence from pyrosequencing indicates that natural variation in animal personality is associated with DRD4 DNA methylation. Mol. Ecol. 2016, 25, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Kisliouk, T.; Meiri, N. A critical role for dynamic changes in histone H3 methylation at the Bdnf promoter during postnatal thermotolerance acquisition. Eur. J. Neurosci. 2009, 30, 1909–1922. [Google Scholar] [CrossRef] [PubMed]
- Yossifoff, M.; Kisliouk, T.; Meiri, N. Dynamic changes in DNA methylation during thermal control establishment affect CREB binding to the brain-derived neurotrophic factor promoter. Eur. J. Neurosci. 2008, 28, 2267–2277. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhi, L.; Liu, Y.; Shen, J.; Liu, L.; Yao, J.; Yang, X. Effect of in ovo feeding of folic acid on the folate metabolism, immune function and epigenetic modification of immune effector molecules of broiler. Br. J. Nutr. 2016, 115, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sun, Q.; Li, X.; Wang, M.; Cai, D.; Li, X.; Zhao, R. In Ovo injection of betaine affects hepatic cholesterol metabolism through epigenetic gene regulation in newly hatched chicks. PLoS ONE 2015, 10, e0122643. [Google Scholar] [CrossRef] [PubMed]
- Laine, V.N.; Gossmann, T.I.; Schachtschneider, K.M.; Garroway, C.J.; Madsen, O.; Verhoeven, K.J.F.; De Jager, V.; Megens, H.J.; Warren, W.C.; Minx, P.; et al. Evolutionary signals of selection on cognition from the great tit genome and methylome. Nat. Commun. 2016, 7, 10474. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Bock, C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012, 13, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef]
- Wu, T.P.; Wang, T.; Seetin, M.G.; Lai, Y.; Zhu, S.; Lin, K.; Liu, Y.; Byrum, S.D.; Mackintosh, S.G.; Zhong, M.; et al. DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature 2016, 532, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P. CpG-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Keshet, I.; Lieman-Hurwitz, J.; Cedar, H. DNA methylation affects the formation of active chromatin. Cell 1986, 44, 535–543. [Google Scholar] [CrossRef]
- Razin, A.; Cedar, H. DNA methylation and gene expression. Microbiol. Rev. 1991, 55, 451–458. [Google Scholar] [PubMed]
- Bird, A. The essentials of DNA methylation. Cell 1992, 70, 5–8. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Busche, S.; Shao, X.; Caron, M.; Kwan, T.; Allum, F.; Cheung, W.A.; Susan Westfall, B.G.; Simon, M.-M. Population whole-genome bisulfite sequencing across two tissues highlights the environment as the principal source of human methylome variation. Genome Biol. 2015, 16, 290. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.L.; Kodell, R.L.; Moore, S.R.; Cooney, C.A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998, 12, 949–957. [Google Scholar] [PubMed]
- Kornberg, R.D.; Lorch, Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 1999, 98, 285–294. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Eberharter, A.; Becker, P.B. Histone acetylation: A switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002, 3, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Lämke, J.; Brzezinka, K.; Altmann, S.; Bäurle, I. A hit-and-run heat shock factor governs sustained histone methylation and transcriptional stress memory. EMBO J. 2016, 35, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Coustham, V.; Li, P.; Strange, A.; Lister, C.; Song, J.; Dean, C. Quantitative modulation of polycomb silencing underlies natural variation in vernalization. Science 2012, 337, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. Making sense of chromatin states. Nat. Methods 2011, 8, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.D.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Stamatoyannopoulos, J.A.; Costello, J.F.; Ren, B.; Milosavljevic, A.; Meissner, A.; Kellis, M.; Marra, M.A.; Beaudet, A.L.; Ecker, J.R.; et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 2010, 28, 1045–1048. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.-B. Perspectives of International Human Epigenome Consortium. Genom. Inform. 2013, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Celniker, S.E.; Dillon, L.A.L.; Gerstein, M.B.; Gunsalus, K.C.; Henikoff, S.; Karpen, G.H.; Kellis, M.; Lai, E.C.; Lieb, J.D.; MacAlpine, D.M.; et al. Unlocking the secrets of the genome. Nature 2009, 459, 927–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, L.; Archibald, A.L.; Bottema, C.D.; Brauning, R.; Burgess, S.C.; Burt, D.W.; Casas, E.; Cheng, H.H.; Clarke, L.; Couldrey, C.; et al. Coordinated international action to accelerate genome-to-phenome with FAANG, the Functional Annotation of Animal Genomes project. Genome Biol. 2015, 16, 57. [Google Scholar] [CrossRef] [PubMed]
- Kurdyukov, S.; Bullock, M. DNA Methylation Analysis: Choosing the Right Method. Biology 2016, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Urich, M.A.; Nery, J.R.; Lister, R.; Schmitz, R.J.; Ecker, J.R. MethylC-seq library preparation for base-resolution whole-genome bisulfite sequencing. Nat. Protoc. 2015, 10, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Smith, Z.D.; Bock, C.; Boyle, P.; Gnirke, A.; Meissner, A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Protoc. 2011, 6, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Soto, J.; Rodriguez-Antolin, C.; Vallespín, E.; de Castro Carpeño, J.; Ibanez de Caceres, I. The impact of next-generation sequencing on the DNA methylation-based translational cancer research. Transl. Res. 2016, 169, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Wang, T.; Coarfa, C.; Nagarajan, R.P.; Hong, C.; Downey, S.L.; Johnson, B.E.; Fouse, S.D.; Delaney, A.; Zhao, Y.; et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotechnol. 2010, 28, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Adams, C.; Landers, M.; Dudas, M.; Krissinger, D.; Marnellos, G.; Bonneville, R.; Xu, M.; Wang, J.; Huang, T.H.M.; et al. High resolution detection and analysis of CpG dinucleotides methylation using MBD-Seq technology. PLoS ONE 2011, 6, e22226. [Google Scholar] [CrossRef] [PubMed]
- Dolinoy, D.C.; Huang, D.; Jirtle, R.L. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. USA 2007, 104, 13056–13061. [Google Scholar] [CrossRef] [PubMed]
- Pértille, F.; Guerrero-Bosagna, C.; Da Silva, V.H.; Boschiero, C.; da Silva Nunes, J.D.R.; Ledur, M.C.; Jensen, P.; Coutinho, L.L. High-throughput and Cost-effective Chicken Genotyping Using Next-Generation Sequencing. Sci. Rep. 2016, 6, 26929. [Google Scholar] [CrossRef] [PubMed]
- Pértille, F.; Brantsæter, M.; Nordgreen, J.; Coutinho, L.L.; Janczak, A.M.; Jensen, P.; Guerrero-Bosagna, C. DNA methylation profiles in red blood cells of adult hens correlate with their rearing conditions. J. Exp. Biol. 2017, 220, 3579–3587. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Ponnaluri, V.K.C.; Ehrlich, K.C.; Zhang, G.; Lacey, M.; Johnston, D.; Pradhan, S.; Ehrlich, M. Association of 5-hydroxymethylation and 5-methylation of DNA cytosine with tissue-specific gene expression. Epigenetics 2017, 12, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Globisch, D.; Münzel, M.; Müller, M.; Michalakis, S.; Wagner, M.; Koch, S.; Brückl, T.; Biel, M.; Carell, T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 2010, 5, e15367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, M.J.; Ost, T.W.; Beraldi, D.; Bell, N.M.; Branco, M.R.; Reik, W.; Balasubramanian, S. Oxidative bisulfite sequencing of 5-methylcytosine and 5-hydroxymethylcytosine. Nat. Protoc. 2013, 8, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.A.; Li, A.X.; Wu, X.; Pfeifer, G.P. Single base resolution analysis of 5-methylcytosine and 5-hydroxymethylcytosine by RRBS and TAB-RRBS. Methods Mol. Biol. 2015, 1238, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Hirst, M.; Bainbridge, M.; Bilenky, M.; Zhao, Y.; Zeng, T.; Euskirchen, G.; Bernier, B.; Varhol, R.; Delaney, A.; et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 2007, 4, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.R.; O’Donnell, A.H.; Rollins, R.A.; Peckham, H.E.; Lee, C.; Milekic, M.H.; Chanrion, B.; Fu, Y.; Su, T.; Hibshoosh, H.; et al. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res. 2010, 20, 972–980. [Google Scholar] [CrossRef] [PubMed]
- David, S.A.; Piégu, B.; Hennequet-Antier, C.; Pannetier, M.; Aguirre-Lavin, T.; Crochet, S.; Bordeau, T.; Couroussé, N.; Brionne, A.; Bigot, Y.; et al. An Assessment of Fixed and Native Chromatin Preparation Methods to Study Histone Post-Translational Modifications at a Whole Genome Scale in Skeletal Muscle Tissue. Biol. Proced. Online 2017, 19, 10. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.S.; Pugh, B.F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 2011, 147, 1408–1419. [Google Scholar] [CrossRef] [PubMed]
- International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [Google Scholar] [CrossRef]
- McQueen, H.A.; Fantes, J.; Cross, S.H.; Clark, V.H.; Archibald, A.L.; Bird, A.P. CpG islands of chicken are concentrated on microchromosomes. Nat. Genet. 1996, 12, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Warren, W.C.; Hillier, L.W.; Tomlinson, C.; Minx, P.; Kremitzki, M.; Graves, T.; Markovic, C.; Bouk, N.; Pruitt, K.D.; Thibaud-Nissen, F.; et al. A New Chicken Genome Assembly Provides Insight into Avian Genome Structure. G3 Genes Genomes Genet. 2017, 7, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Dalloul, R.A.; Long, J.A.; Zimin, A.V.; Aslam, L.; Beal, K.; Blomberg, L.A.; Bouffard, P.; Burt, D.W.; Crasta, O.; Crooijmans, R.P.; et al. Multi-platform next-generation sequencing of the domestic turkey (Meleagris gallopavo): Genome assembly and analysis. PLoS Biol. 2010, 8, e1000475. [Google Scholar] [CrossRef] [PubMed]
- NCBI. Eukaryotic Genomes Annotated at NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome/annotation_euk/all/ (accessed on 6 July 2017).
- Li, Q.; Li, N.; Hu, X.; Li, J.; Du, Z.; Chen, L.; Yin, G.; Duan, J.; Zhang, H.; Zhao, Y.; et al. Genome-wide mapping of DNA methylation in chicken. PLoS ONE 2011, 6, e19428. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Xu, H.; Li, Z.; Zheng, X.; Jia, X.; Nie, Q.; Zhang, X. Comparison of the genome-wide DNA methylation profiles between fast-growing and slow-growing broilers. PLoS ONE 2013, 8, e56411. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zhang, Y.; Sun, D.; Wang, Y.; Yu, Y. Analysis on DNA methylation of various tissues in chicken. Anim. Biotechnol. 2007, 18, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, R.; Wang, Y.; Hu, X.; Zhao, Y.; Li, L.; Feng, C.; Gu, X.; Liang, F.; Lamont, S.J.; et al. Genome-wide DNA methylome variation in two genetically distinct chicken lines using MethylC-seq. BMC Genom. 2015, 16, 851. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Zhan, F.; VanderKraats, N.D.; Hiken, J.F.; Edwards, J.R.; Zhang, H.; Zhao, K.; Song, J. DNMT gene expression and methylome in Marek’s disease resistant and susceptible chickens prior to and following infection by MDV. Epigenetics 2013, 8, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Gou, Z.; Liu, R.; Zhao, G.; Zheng, M.; Li, P.; Wang, H.; Zhu, Y.; Chen, J.; Wen, J. Epigenetic modification of TLRs in leukocytes is associated with increased susceptibility to Salmonella enteritidis in chickens. PLoS ONE 2012, 7, e33627. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yan, F.B.; Li, F.; Jiang, K.R.; Li, D.H.; Han, R.L.; Li, Z.J.; Jiang, R.R.; Liu, X.J.; Kang, X.T.; et al. Genome-wide DNA methylation profiles reveal novel candidate genes associated with meat quality at different age stages in hens. Sci. Rep. 2017, 7, 45564. [Google Scholar] [CrossRef] [PubMed]
- Derks, M.F.L.; Schachtschneider, K.M.; Madsen, O.; Schijlen, E.; Verhoeven, K.J.F.; van Oers, K. Gene and transposable element methylation in great tit (Parus major) brain and blood. BMC Genom. 2016, 17, 332. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Mitra, A.; Tian, F.; Chang, S.; Zhang, H.; Cui, K.; Yu, Y.; Zhao, K.; Song, J. Histone methylation analysis and pathway predictions in chickens after MDV infection. PLoS ONE 2012, 7, e41849. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Luo, J.; Zhang, H.; Cui, K.; Zhao, K.; Song, J. Marek’s disease virus infection induces widespread differential chromatin marks in inbred chicken lines. BMC Genom. 2012, 13, 557. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Luo, J.; He, Y.; Gu, Y.; Zhang, H.; Zhao, K.; Cui, K.; Song, J. Histone modifications induced by MDV infection at early cytolytic and latency phases. BMC Genom. 2015, 16, 311. [Google Scholar] [CrossRef] [PubMed]
- Jahan, S.; Xu, W.; He, S.; Gonzalez, C.; Delcuve, G.P.; Davie, J.R. The chicken erythrocyte epigenome. Epigenet. Chromatin 2016, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Luo, J.; Mitra, A.; Chang, S.; Tian, F.; Zhang, H.; Yuan, P.; Zhou, H.; Song, J. Temporal transcriptome changes induced by MDV in Marek’s disease-resistant and -susceptible inbred chickens. BMC Genom. 2011, 12, 501. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.N.; Vierbuchen, T.; Hemberg, M.; Rubin, A.A.; Ling, E.; Couch, C.H.; Stroud, H.; Spiegel, I.; Farh, K.K.H.; Harmin, D.A.; et al. Genome-wide identification and characterization of functional neuronal activity-dependent enhancers. Nat. Neurosci. 2014, 17, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Boleli, I.; Morita, V.; Matos, J., Jr.; Thimotheo, M.; Almeida, V. Poultry Egg Incubation: Integrating and Optimizing Production Efficiency. Rev. Bras. Ciência Avícola 2016, 18. [Google Scholar] [CrossRef]
- Rigaill, G.; Balzergue, S.; Brunaud, V.; Blondet, E.; Rau, A.; Rogier, O.; Caius, J.; Maugis-Rabusseau, C.; Soubigou-Taconnat, L.; Aubourg, S.; et al. Synthetic data sets for the identification of key ingredients for RNA-seq differential analysis. Brief. Bioinform. 2016. [Google Scholar] [CrossRef] [PubMed]
- Daxinger, L.; Whitelaw, E. Transgenerational epigenetic inheritance: More questions than answers. Genome Res. 2010, 20, 1623–1628. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Gu, H.; Bock, C.; Gnirke, A.; Meissner, A. High-throughput bisulfite sequencing in mammalian genomes. Methods 2009, 48, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Hansen, K.D.; Meissner, A.; Arye, M.J. Coverage recommendations for methylation analysis by whole-genome bisulfite sequencing. Nat. Methods 2015, 12, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Yong, W.S.; Hsu, F.M.; Chen, P.Y. Profiling genome-wide DNA methylation. Epigenet. Chromatin 2016, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Stein, L. Genome annotation: From sequence to biology. Nat. Rev. Genet. 2001, 2, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Doherty, R.; Couldrey, C. Exploring genome wide bisulfite sequencing for DNA methylation analysis in livestock: A technical assessment. Front. Genet. 2014, 5, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landt, S.G.; Marinov, G.K.; Kundaje, A.; Kheradpour, P.; Pauli, F.; Batzoglou, S.; Bernstein, B.E.; Bickel, P.; Brown, J.B.; Cayting, P.; et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012, 22, 1813–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.L.; Luquette, L.J.; Ho, J.W.K.; Ferrari, F.; Tolstorukov, M.; Minoda, A.; Issner, R.; Epstein, C.B.; Karpen, G.H.; Kuroda, M.I.; et al. Impact of sequencing depth in ChIP-seq experiments. Nucleic Acids Res. 2014, 42, e74. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; He, X. Integrating Epigenomics into the Understanding of Biomedical Insight. Bioinform. Biol. Insights 2016, 10, 267–289. [Google Scholar] [CrossRef] [PubMed]
- The ENCODE Project Consortium. Whole-Genome Bisulfite Sequencing Data Standards and Processing Pipeline. Available online: https://www.encodeproject.org/wgbs/ (accessed on 6 July 2017).
- Epigenome Blueprint. ChIP-Seq Analysis Pipeline. Available online: http://dcc.blueprint-epigenome.eu/#/md/chip_seq_grch37 (accessed on 6 July 2017).
- Sun, Z.; Cunningham, J.; Slager, S.; Kocher, J.P. Base resolution methylome profiling: Considerations in platform selection, data preprocessing and analysis. Epigenomics 2015, 7, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Ewing, B.; Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Del Fabbro, C.; Scalabrin, S.; Morgante, M.; Giorgi, F.M. An extensive evaluation of read trimming effects on Illumina NGS data analysis. PLoS ONE 2013, 8, e85024. [Google Scholar] [CrossRef] [PubMed]
- Schbath, S.; Martin, V.; Zytnicki, M.; Fayolle, J.; Loux, V.; Gibrat, J.F. Mapping reads on a genomic sequence: An algorithmic overview and a practical comparative analysis. J. Comput. Biol. 2012, 19, 796–813. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Sun, S. Comparing five statistical methods of differential methylation identification using bisulfite sequencing data. Stat. Appl. Genet. Mol. Biol. 2016, 15, 173–191. [Google Scholar] [CrossRef] [PubMed]
- Steinhauser, S.; Kurzawa, N.; Eils, R.; Herrmann, C. A comprehensive comparison of tools for differential ChIP-seq analysis. Brief. Bioinform. 2016, 17, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Belton, J.M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
David, S.-A.; Mersch, M.; Foissac, S.; Collin, A.; Pitel, F.; Coustham, V. Genome-Wide Epigenetic Studies in Chicken: A Review. Epigenomes 2017, 1, 20. https://doi.org/10.3390/epigenomes1030020
David S-A, Mersch M, Foissac S, Collin A, Pitel F, Coustham V. Genome-Wide Epigenetic Studies in Chicken: A Review. Epigenomes. 2017; 1(3):20. https://doi.org/10.3390/epigenomes1030020
Chicago/Turabian StyleDavid, Sarah-Anne, Marjorie Mersch, Sylvain Foissac, Anne Collin, Frédérique Pitel, and Vincent Coustham. 2017. "Genome-Wide Epigenetic Studies in Chicken: A Review" Epigenomes 1, no. 3: 20. https://doi.org/10.3390/epigenomes1030020