
Mining Novel Candidate Imprinted Genes Using Genome-Wide Methylation Screening and Literature Review

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Compilation of Known Imprinted Genes in Human and Mice
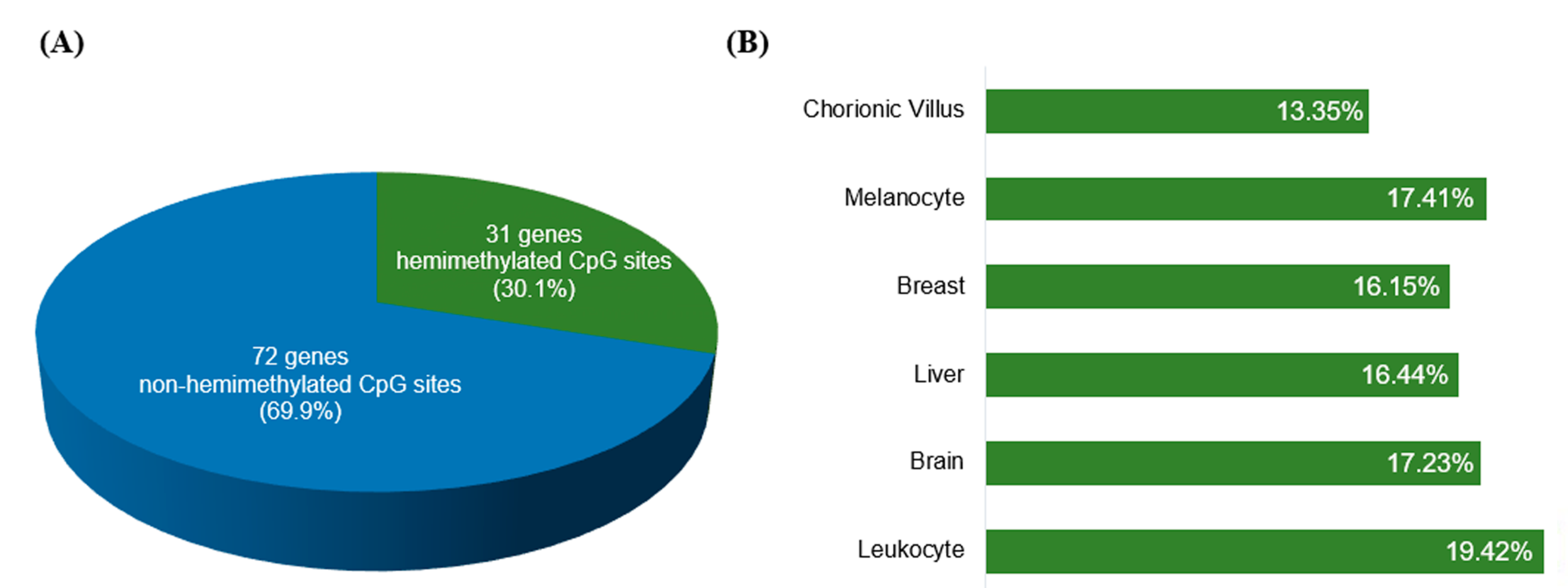
2.2. Methylation Array Data Processing and Selection Criteria of IGs
2.3. Known Imprinted Gene Cpgs Retrieved from the HM450K Methylation Data
2.4. Mining Novel Candidate Imprinted Genes Using the HM450K Methylation Data
2.5. Characterization of Human Orthologues of Known Mouse IGs, Using the HM450K Human Methylation Data
2.6. Comparison of Known and Candidate IGs Detected in Human Imprinting Studies Using High-Throughput Technologies
3. Discussion
4. Materials and Methods
4.1. Human Tissue Samples
4.2. Genome-Wide DNA Methylation Analysis
4.3. Array Quality Control and Data Processing
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chess, A. Mechanisms and consequences of widespread random monoallelic expression. Nat. Rev. Genet. 2012, 13, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Henckel, A.; Arnaud, P. Genome-wide identification of new imprinted genes. Brief. Funct. Genom. 2010, 9, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Morison, I.M.; Ramsay, J.P.; Spencer, H.G. A census of mammalian imprinting. Trends Genet. 2005, 21, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Walter, J. Genomic imprinting: parental influence on the genome. Nat. Rev. Genet. 2001, 2, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.; Reik, W. How imprinting centres work. Cytogenet. Genome Res. 2006, 113, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Royo, H.; Cavaillé, J. Non-coding RNAs in imprinted gene clusters. Biol. Cell 2008, 100, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Bartolomei, M.S. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell 2013, 152, 1308–1323. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.A.; Ferguson-Smith, A.C. Mechanisms regulating imprinted genes in clusters. Curr. Opin. Cell Biol. 2007, 19, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.R.; Bartolomei, M.S. Chromatin regulators of genomic imprinting. Biochim. Biophys. Acta-Gene Regul. Mech. 2014, 1839, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Yuen, R.K.; Jiang, R.; Peñaherrera, M.S.; McFadden, D.E.; Robinson, W.P. Genome-wide mapping of imprinted differentially methylated regions by DNA methylation profiling of human placentas from triploidies. Epigenet. Chromatin 2011, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Choufani, S.; Shapiro, J.S.; Susiarjo, M.; Butcher, D.T.; Grafodatskaya, D.; Lou, Y.; Ferreira, J.C.; Pinto, D.; Scherer, S.W.; Shaffer, L.G.; et al. A novel approach identifies new differentially methylated regions (DMRs) associated with imprinted genes. Genome Res. 2011, 21, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, K.; Trujillo, A.M.; Tayama, C.; Camprubi, C.; Yoshida, W.; Lapunzina, P.; Sanchez, A.; Soejima, H.; Aburatani, H.; Nagae, G.; et al. Methylation screening of reciprocal genome-wide UPDs identifies novel human-specific imprinted genes. Hum. Mol. Genet. 2011, 20, 3188–3197. [Google Scholar] [CrossRef] [PubMed]
- Barbaux, S.; Gascoin-Lachambre, G.; Buffat, C.; Monnier, P.; Mondon, F.; Tonanny, M.-B.; Pinard, A.; Auer, J.; Bessières, B.; Barlier, A.; et al. A genome-wide approach reveals novel imprinted genes expressed in the human placenta. Epigenetics 2012, 7, 1079–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Docherty, L.E.; Rezwan, F.I.; Poole, R.L.; Jagoe, H.; Lake, H.; Lockett, G.A.; Arshad, H.; Wilson, D.I.; Holloway, J.W.; Temple, I.K.; et al. Genome-wide DNA methylation analysis of patients with imprinting disorders identifies differentially methylated regions associated with novel candidate imprinted genes. J. Med. Genet. 2014, 51, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Court, F.; Tayama, C.; Romanelli, V.; Martin-Trujillo, A.; Iglesias-Platas, I.; Okamura, K.; Sugahara, N.; Simon, C.; Moore, H.; Harness, J.V.; et al. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014, 24, 554–569. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Viltrop, T.; Tiirats, A.; Rajashekar, B.; Reimann, E.; Kõks, S.; Rull, K.; Milani, L.; Acharya, G.; Basnet, P.; et al. Using RNA sequencing for identifying gene imprinting and random monoallelic expression in human placenta. Epigenetics 2014, 9, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Steyaert, S.; Van Criekinge, W.; De Paepe, A.; Denil, S.; Mensaert, K.; Vandepitte, K.; Vanden Berghe, W.; Trooskens, G.; De Meyer, T. SNP-guided identification of monoallelic DNA-methylation events from enrichment-based sequencing data. Nucleic Acids Res. 2014, 42, e157. [Google Scholar] [CrossRef] [PubMed]
- Babak, T.; DeVeale, B.; Tsang, E.K.; Zhou, Y.; Li, X.; Smith, K.S.; Kukurba, K.R.; Zhang, R.; Li, J.B.; van der Kooy, D.; et al. Genetic conflict reflected in tissue-specific maps of genomic imprinting in human and mouse. Nat. Genet. 2015, 47, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Baran, Y.; Subramaniam, M.; Biton, A.; Tukiainen, T.; Tsang, E.K.; Rivas, M.A.; Pirinen, M.; Gutierrez-Arcelus, M.; Smith, K.S.; Kukurba, K.R.; et al. The landscape of genomic imprinting across diverse adult human tissues. Genome Res. 2015, 25, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Catalogue of Imprinted Genes and Parent-of-Origin Effects in Humans and Animals. Available online: http://igc.otago.ac.nz/home.html (accessed on 27 July 2017).
- Geneimprint. Available online: http://www.geneimprint.com/ (accessed on 3 August 2017).
- Wang, X.; Clark, A.G. Using next-generation RNA sequencing to identify imprinted genes. Heredity 2014, 113, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Woodfine, K.; Huddleston, J.E.; Murrell, A. Quantitative analysis of DNA methylation at all human imprinted regions reveals preservation of epigenetic stability in adult somatic tissue. Epigenet. Chromatin 2011, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjornsson, H.; Albert, T. SNP-specific array-based allele-specific expression analysis. Genome Res. 2008, 18, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Tarutani, Y.; Takayama, S. Monoallelic gene expression and its mechanisms. Curr. Opin. Plant Biol. 2011, 14, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Abramowitz, L.K.; Bartolomei, M.S. Genomic imprinting: Recognition and marking of imprinted loci. Curr. Opin. Genet. Dev. 2012, 22, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Pant, P.V.K.; Tao, H.; Beilharz, E.J.; Ballinger, D.G.; Cox, D.R.; Frazer, K.A. Analysis of allelic differential expression in human white blood cells. Genome Res. 2006, 16, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Maynard, N.D.; Chen, J.; Stuart, R.K.; Fan, J.B.; Ren, B. Genome-wide mapping of allele-specific protein-DNA interactions in human cells. Nat. Methods 2008, 5, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Pollard, K.S.; Serre, D.; Wang, X.; Tao, H.; Grundberg, E.; Hudson, T.J.; Clark, A.G.; Frazer, K. A genome-wide approach to identifying novel-imprinted genes. Hum. Genet. 2008, 122, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.; Hong, C.; Xing, X.; Zhou, X.; Li, D.; Coarfa, C.; Bell, R.J.A.; Maire, C.L.; Ligon, K.L.; Sigaroudinia, M.; et al. Intermediate DNA methylation is a conserved signature of genome regulation. Nat. Commun. 2015, 6, 6363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Maniatis, T. A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell 1999, 97, 779–790. [Google Scholar] [CrossRef]
- Esumi, S.; Kakazu, N.; Taguchi, Y.; Hirayama, T.; Sasaki, A.; Hirabayashi, T.; Koide, T.; Kitsukawa, T.; Hamada, S.; Yagi, T. Monoallelic yet combinatorial expression of variable exons of the protocadherin-alpha gene cluster in single neurons. Nat. Genet. 2005, 37, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, R.; Kato, H.; Kawamura, Y.; Esumi, S.; Hirayama, T.; Hirabayashi, T.; Yagi, T. Allelic gene regulation of Pcdh-α and Pcdh-γ clusters involving both monoallelic and biallelic expression in single Purkinje cells. J. Biol. Chem. 2006, 281, 30551–30560. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Kaneko, R.; Izawa, T.; Kawaguchi, M.; Kitsukawa, T.; Yagi, T. Single-neuron diversity generated by Protocadherin-β cluster in mouse central and peripheral nervous systems. Front. Mol. Neurosci. 2012, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, S.; Kawaguchi, M.; Kobayashi, T.; Tarusawa, E.; Toyama, T.; Okano, M.; Oda, M.; Nakauchi, H.; Yoshimura, Y.; Sanbo, M.; et al. Developmental epigenetic modification regulates stochastic expression of clustered protocadherin genes, generating single neuron diversity. Neuron 2014, 82, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Gendrel, A.V.; Tang, Y.A.; Suzuki, M.; Godwin, J.; Nesterova, T.B.; Greally, J.M.; Heard, E.; Brockdorff, N. Epigenetic functions of Smchd1 repress gene clusters on the inactive X chromosome and on autosomes. Mol. Cell Biol. 2013, 33, 3150–3165. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, J.M.; Higgins, M.J.; Gebuhr, T.C.; Shows, T.B.; Saitoh, S.; Nicholls, R.D. A model system to study genomic imprinting of human genes. Proc. Natl. Acad. Sci. USA 1998, 95, 14857–14862. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.M.; Udayashankar, R.; Moore, H.D.; Moore, G.E. Telomeric NAP1L4 and OSBPL5 of the KCNQ1 cluster, and the DECORIN gene are not imprinted in human trophoblast stem cells. PLoS ONE 2010, 5, e11595. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Noskov, V.N.; Lu, X.; Bergmann, A.; Ren, X.; Warth, T.; Richardson, P.; Kouprina, N.; Stubbs, L. Discovery of a novel, paternally expressed ubiquitin-specific processing protease gene through comparative analysis of an imprinted region of mouse chromosome 7 and human chromosome 19q13.4. Genome Res. 2000, 10, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bergmann, A.; Wehri, E.; Lu, X.; Stubbs, L. Imprinting and evolution of two Kruppel-type zinc-finger genes, ZIM3 and ZNF264, located in the PEG3/USP29 imprinted domain. Genomics 2001, 77, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.M.; Kim, J. DNA methylation analysis of the mammalian PEG3 imprinted domain. Gene 2009, 442, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.J.; Migliavacca, E.; Dupre, Y.; Stathaki, E.; Sailani, M.R.; Baumer, A.; Schinzel, A.; Mackay, D.J.; Robinson, D.O.; Cobellis, G.; et al. Methylation profiling in individuals with uniparental disomy identifies novel differentially methylated regions on chromosome 15. Genome Res. 2010, 20, 1271–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, O.; McNoe, L.A.; Eccles, M.R.; Morison, I.M. Reeve AEHuman insulin-like growth factor type I and type II receptors are not imprinted. Hum. Mol. Genet. 1993, 2, 2163–2165. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, L.T.; Lucena Ferretti, R.E.; Farfel, J.M.; Leite, R.; Pasqualucci, C.A.; Rosemberg, S.; Nitrini, R.; Saldiva, P.H.N.; Jacob Filho, W. Brain bank of the Brazilian aging brain study group-A milestone reached and more than 1,600 collected brains. Cell Tissue Bank. 2007, 8, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Type | Location | Number of Shared CpGs (>2 Tissues) | Previously Reported Evidence of Imprinting | Ref. |
|---|---|---|---|---|---|
| PLEC | protein-coding | 8q24.3 | 7 | Candidate locus with three hypomethylated probes in BWS patients with multilocus hypomethylation (HM450K) | [15] |
| PTCHD3 | protein-coding | 10p12.1 | 7 | Candidate locus with four hypomethylated probes in BWS patients with multilocus hypomethylation (HM450K) | [15] |
| COX4I2 | protein-coding | 20q11.21 | 6 | ||
| FOXR1 | protein-coding | 11q23.3 | 6 | ||
| ZNF331 a | protein-coding | 19q13.42 | 6 | Validated isoform-specific imprinting | [15,16,19,20] |
| CCDC144NL | protein-coding | 17p11.2 | 4 | ||
| DEF8 | protein-coding | 16q24.3 | 4 | ||
| H3F3C | protein-coding | 12p11.21 | 4 | ||
| KAZALD1 | protein-coding | 10q24.31 | 4 | ||
| KIAA2013 | protein-coding | 1p36.22 | 4 | Candidate locus with two hypomethylated probes in BWS patients with multilocus hypomethylation (HM450K) | [15] |
| LOC494141 | Pseudogene | 11p15.1 | 4 | ||
| SYCE1 | protein-coding | 10q26.3 | 4 | Candidate locus with two probes mapping to both SYCE1/SPRNP1 (HM27K); ASE demonstrated in multiples human tissues | [12,19] |
| CCDC144A | protein-coding | 17p11.2 | 3 | ||
| CECR7 | ncRNA | 22q11.1 | 3 | ||
| CENPF | protein-coding | 1q41 | 3 | ||
| FANK1 | protein-coding | 10q26.2 | 3 | ||
| ANKRD36BP2 | Pseudogene | 2p11.2 | 3 | ||
| HTR5A | protein-coding | 7q36.1 | 3 | Candidate locus with six probes with maternal methylation (HM450K) | [16] |
| TONSL | protein-coding | 8q24.3 | 3 | ||
| PDE6B | protein-coding | 4p16.3 | 3 | ||
| SLC16A3 | protein-coding | 17q25 | 3 | ||
| SLC25A2 | protein-coding | 5q31 | 3 | ||
| SRMS | protein-coding | 20q13.33 | 3 | ||
| TCEA2 | protein-coding | 20q13.33 | 3 | ||
| TCL1A | protein-coding | 14q32.13 | 3 | ||
| VPS28 | protein-coding | 8q24.3 | 3 | ||
| ZNF232 | protein-coding | 17p13.2 | 3 | Candidate locus with one probe with maternal methylation exclusively in placenta (HM27K) | [13] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonaldi, A.; Kashiwabara, A.; Araújo, É.S.d.; Pereira, L.V.; Paschoal, A.R.; Andozia, M.B.; Villela, D.; Rivas, M.P.; Suemoto, C.K.; Pasqualucci, C.A.; et al. Mining Novel Candidate Imprinted Genes Using Genome-Wide Methylation Screening and Literature Review. Epigenomes 2017, 1, 13. https://doi.org/10.3390/epigenomes1020013
Bonaldi A, Kashiwabara A, Araújo ÉSd, Pereira LV, Paschoal AR, Andozia MB, Villela D, Rivas MP, Suemoto CK, Pasqualucci CA, et al. Mining Novel Candidate Imprinted Genes Using Genome-Wide Methylation Screening and Literature Review. Epigenomes. 2017; 1(2):13. https://doi.org/10.3390/epigenomes1020013
Chicago/Turabian StyleBonaldi, Adriano, André Kashiwabara, Érica S.de Araújo, Lygia V. Pereira, Alexandre R. Paschoal, Mayra B. Andozia, Darine Villela, Maria P. Rivas, Claudia K. Suemoto, Carlos A. Pasqualucci, and et al. 2017. "Mining Novel Candidate Imprinted Genes Using Genome-Wide Methylation Screening and Literature Review" Epigenomes 1, no. 2: 13. https://doi.org/10.3390/epigenomes1020013