
Genetic Differentiation of the Bloodsucking Midge Forcipomyia taiwana (Diptera: Ceratopogonidae): Implication of the Geographic Isolation by the Central Mountain Ranges in Taiwan
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
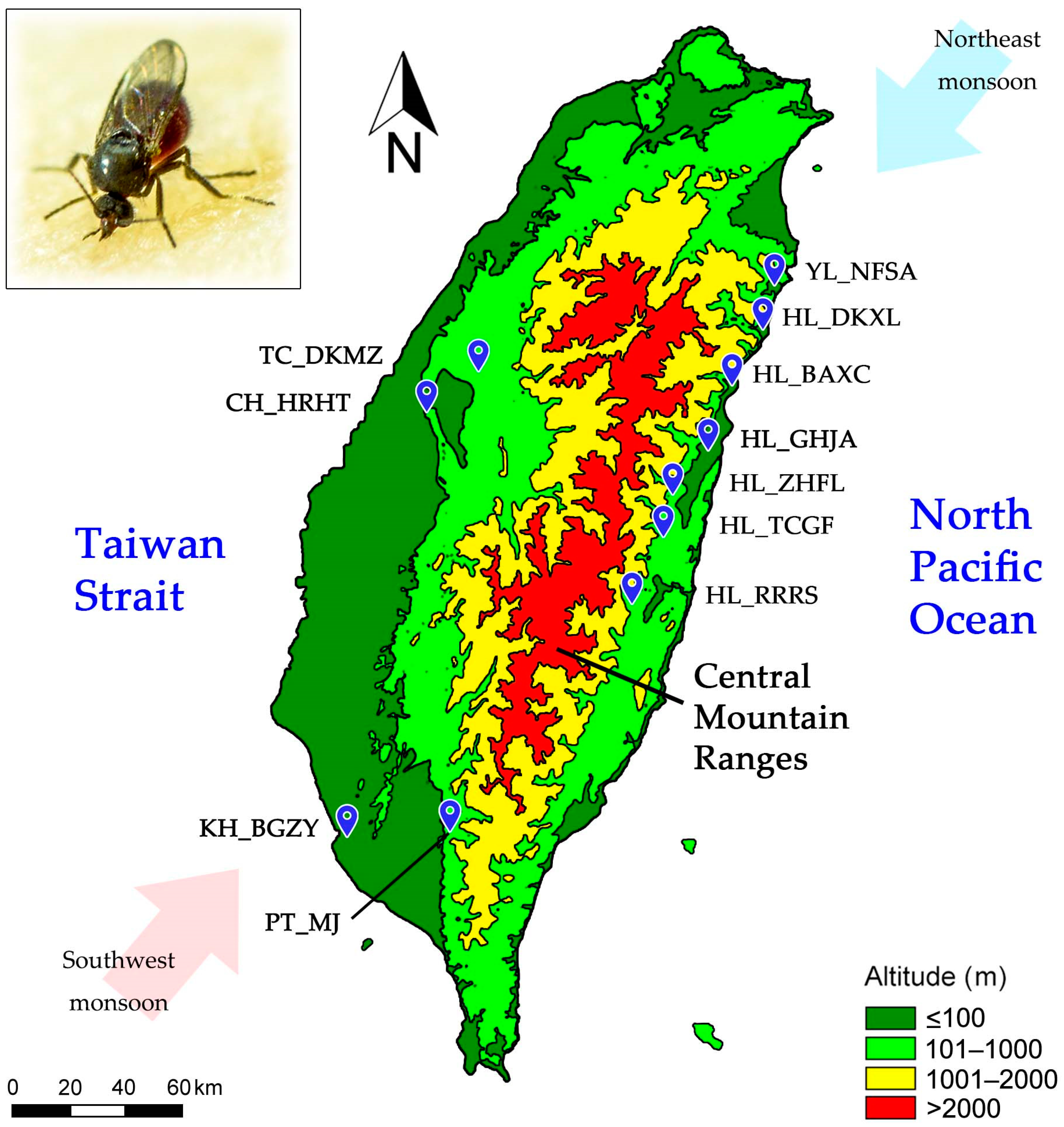
2.1. Sample Collection
2.2. Isolation of Genomic DNA
2.3. Amplification of Microsatellite Markers and Fragment Analysis
2.4. Genotype Scoring and Data Analysis
3. Results
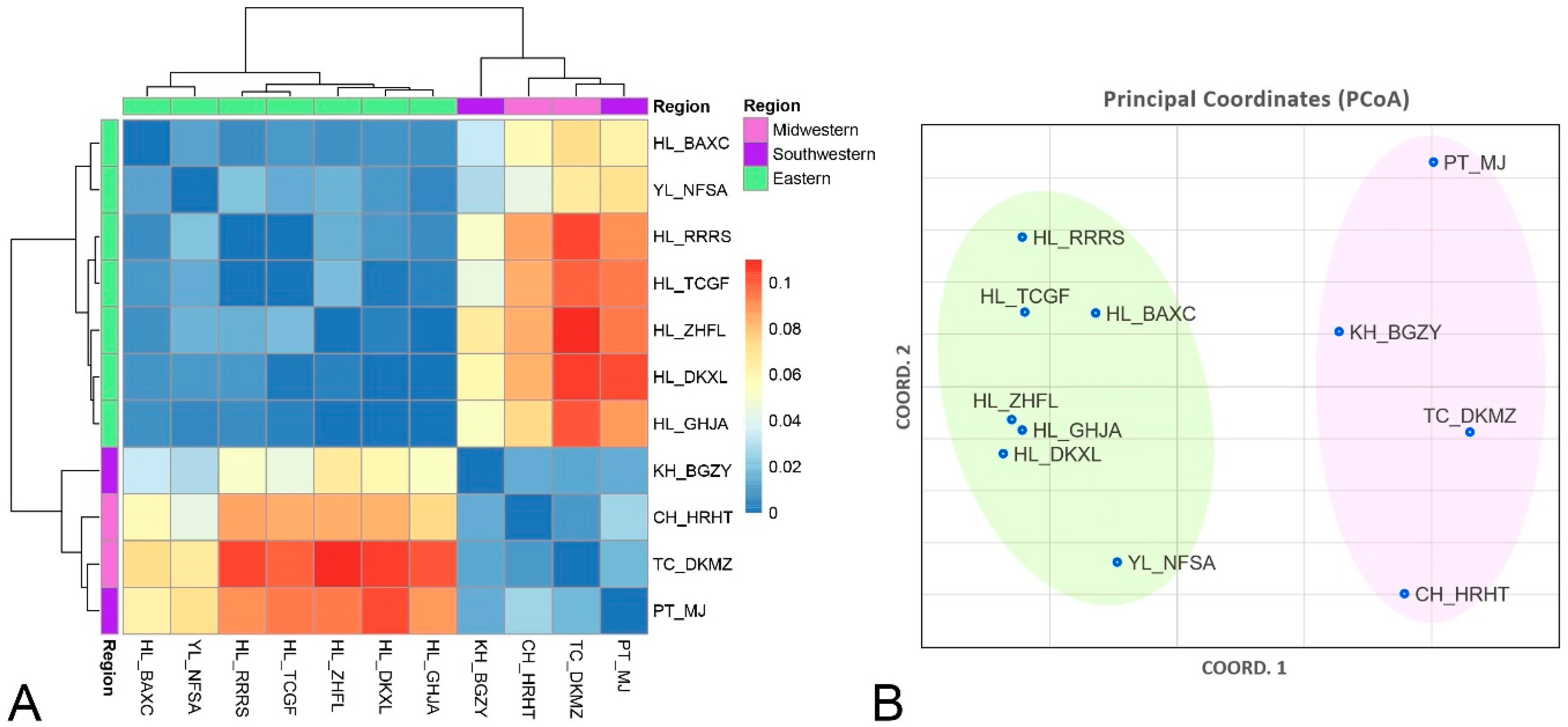
3.1. Genetic Structures of F. taiwana Populations Revealed by Microsatellite Analysis
3.2. Regional Genetic Differentiation in F. taiwana across Taiwan’s Central Mountain Range by the Analysis of Molecular Variance
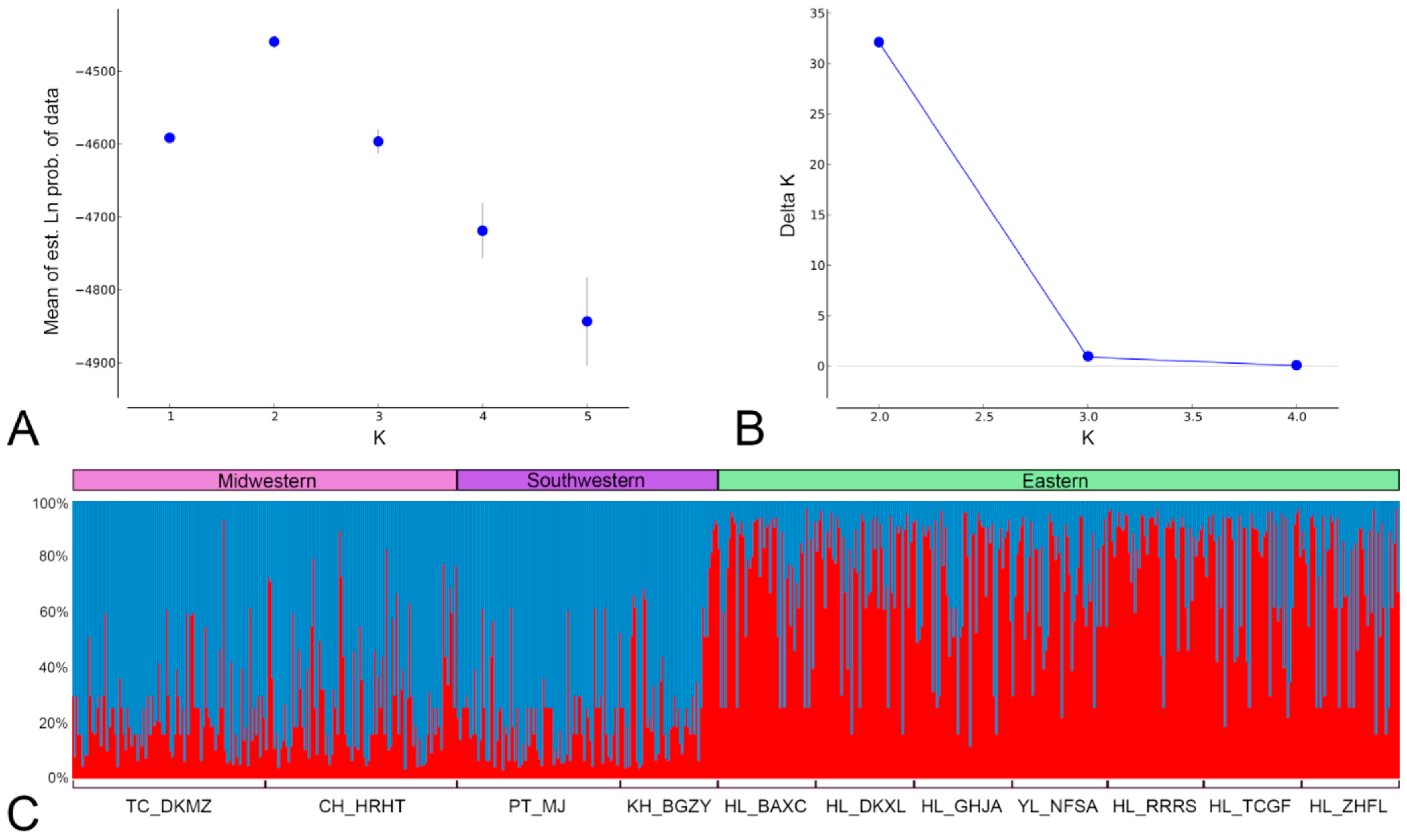
3.3. Bayesian Clustering Analysis of F. taiwana Unveiling Two Distinct Genetic Clusters across Taiwan’s Central Mountain Range
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chuang, Y.Y.; Lin, C.S.; Wang, C.H.; Yeh, C.C. Distribution and seasonal occurrence of Forcipomyia taiwana (Diptera: Ceratopogonidae) in the Nantou area in Taiwan. J. Med. Entomol. 2000, 37, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Lien, J.C.; Lin, Y.N.; Hsu, S.J. The diurnal biting pattern of a bloodsucking midge Forcipomyia (Lasiohelea) taiwana (Shiraki) (Diptera, Ceratopogonidae). Chin. J. Microbiol. Immunol. 1981, 14, 54–56. [Google Scholar]
- Shih, C.L.; Liao, Q.M.; Wang, Y.Y.; Tu, W.C. Abundance and host-seeking activity of the biting midge, Forcipomyia taiwana (Diptera: Ceratopogonidae). J. Asia Pac. Entomol. 2019, 22, 1053–1059. [Google Scholar] [CrossRef]
- Wang, S.C.; Ching, Y.H.; Krishnaraj, P.; Chen, G.Y.; Radhakrishnan, A.S.; Lee, H.M.; Tu, W.C.; Lin, M.D. Oogenesis of Hematophagous Midge Forcipomyia taiwana (Diptera: Ceratopogonidae) and Nuage Localization of Vasa in Germline Cells. Insects 2020, 11, 106. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Hwang, G.Y.; Chen, P.C.; Tu, W.C.; Lee, M.F. Molecular cloning and immunologic characterization of for t 2: A major allergen from the biting midge Forcipomyia taiwana. Allergy 2011, 66, 703–705. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, T. Investigation on general injurious insects. Taiwan. Sotokufu Noji Shikenjo Tolubetsu Hokodu 1913, 8, 286–297. [Google Scholar]
- Yeh, W.B.; Lee, H.M.; Tu, W.C.; Tang, L.C.; Lee, P.Y. Molecular differentiation and diversity of Forcipomyia taiwana (Diptera: Ceratopogonidae) based on the mitochondrial cytochrome oxidase II sequence. J. Med. Entomol. 2009, 46, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Lin, C.N.; Chung, C.L.; Hung, H. Preliminary observations on the larval breeding sites and adult resting places of a bloodsucking midge, Forcipomyia (Lasiohelea) taiwana (Shiraki) (Diptera: Ceratopogonidae). Nat’l Chung Hsing Univ. Bull. Soc. Entomol. 1979, 14, 51–59. [Google Scholar]
- Hsieh, P.Y. Studies on the oviposition, population dynamics and the susceptibility to insect growth regulators of biting midge. In Forcipomyia Taiwana (Shiraki) (Diptera: Ceratopogonidae); National Chung Hsing University: Taichung, Taiwan, 2007. [Google Scholar]
- Shih, C.L.; Tu, W.C. Blood-sucking behavior of female Forcipomyia taiwana (Shiraki) (Diptera: Ceratopogonidae). Formos. Entomol. 2019, 39, 105–114. [Google Scholar]
- Sun, W.K.C. Study of a biting midge, Forcipomyia (Lasiohelea) taiwana (Shiraki) (Diptera: Ceratopogonidae). I. Description of the complete life cycle of the midge reared in the laboratory. Biol. Bull. Tunghai Univ. Taiwan. Taichung 1967, 29, 1–10. [Google Scholar]
- Yeh, C.C.; Chuang, Y.Y. Colonization and bionomics of Forcipomyia taiwana (Diptera:Ceratopogonidae) in the laboratory. J. Med. Entomol. 1996, 33, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.P. Establishing and maintaining colonies of Forcipomyia taiwana in the laboratory. J. Vector Ecol. 2018, 43, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y. (Ed.) Catalogue and Keys of Chinese Ceratopogonidae (Insecta, Diptera); Military Medical Science Press: Beijing, China, 2005; p. 187. [Google Scholar]
- Meeker, N.D.; Hutchinson, S.A.; Ho, L.; Trede, N.S. Method for isolation of PCR-ready genomic DNA from zebrafish tissues. Biotechniques 2007, 43, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Oetting, W.S.; Lee, H.K.; Flanders, D.J.; Wiesner, G.L.; Sellers, T.A.; King, R.A. Linkage analysis with multiplexed short tandem repeat polymorphisms using infrared fluorescence and M13 tailed primers. Genomics 1995, 30, 450–458. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 356–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Meirmans, P.G. Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 2006, 60, 2399–2402. [Google Scholar] [CrossRef] [PubMed]
- Toda, M.; Nishida, M.; Matsui, M.; Lue, K.-Y.; Ota, H. Genetic variation in the Indian rice frog, Rana limnocharis (Amphibia: Anura), in Taiwan, as revealed by allozyme data. Herpetologica 1998, 54, 73–82. [Google Scholar]
- Shih, H.T.; Hung, H.C.; Schubart, C.D.; Chen, C.A.; Chang, H.W. Intraspecific genetic diversity of the endemic freshwater crab Candidiopotamon rathbunae (Decapoda, Brachyura, Potamidae) reflects five million years of the geological history of Taiwan. J. Biogeogr. 2006, 33, 980–989. [Google Scholar] [CrossRef]
- Song, H.; Buhay, J.E.; Whiting, M.F.; Crandall, K.A. Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proc. Natl. Acad. Sci. USA 2008, 105, 13486–13491. [Google Scholar] [CrossRef]
- Zhang, D.X.; Hewitt, G.M. Nuclear integrations: Challenges for mitochondrial DNA markers. Trends Ecol. Evol. 1996, 11, 247–251. [Google Scholar] [CrossRef]
- Glenn, T.C.; Schable, N.A. Isolating microsatellite DNA loci. Methods Enzymol. 2005, 395, 202–222. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Geographic Location in Taiwan (Code) | Geographic Region | Latitude | Longitude | Altitude (Meters) | Sample Size |
|---|---|---|---|---|---|
| Nan’ao Farm, Su’ao Township, Yilan County (YL_NFSA) | Eastern | 24.453 | 121.790 | 18 | 47 |
| Taiwan Cement DAKA Park, Xiulin Township, Hualien County (HL_DKXL) | Eastern | 24.300 | 121.751 | 13 | 48 |
| Bo’ai Road, Xincheng Township, Hualien County (HL_BAXC) | Eastern | 24.126 | 121.651 | 18 | 47 |
| Guanghua 10th Street, Ji’an Township, Hualien County (HL_GHJA) | Eastern | 23.932 | 121.558 | 54 | 48 |
| Zhonghua Road, Fenglin Town, Hualien County (HL_ZHFL) | Eastern | 23.747 | 121.451 | 100 | 48 |
| Tangchang Street, Guangfu Township, Hualien County (HL_TCGF) | Eastern | 23.658 | 121.420 | 121 | 48 |
| Ruisui Ranch, Ruisui Township, Hualien County (HL_RRRS) | Eastern | 23.479 | 121.345 | 154 | 47 |
| Dakeng Scenic Area, Minzheng Li, Taichung City (TC_DKMZ) | Midwestern | 24.172 | 120.752 | 205 | 95 |
| Hushan Rock, Huatan Township, Changhua County (CH_HRHT) | Midwestern | 24.043 | 120.559 | 56 | 94 |
| Majia Township, Pingtung Country (PT_MJ) | Southwestern | 22.703 | 120.648 | 228 | 81 |
| Native Botanical Garden, Zuoying District, Kaohsiung City (KH_BGZY) | Southwestern | 22.680 | 120.301 | 9 | 48 |
| Locus | Size Range (bps) | Motif Type 1 | Primer Sequences 2 and the Concentration of the Primers Used in the PCR Reactions | Number of Alleles | Accession Number |
|---|---|---|---|---|---|
| P1 | 138~183 | (TAA)7 | F: TCAGGCTCGTACGGTTTACT (200 nM) | 7 | MW921469 |
| R: GGAAGTCAAAATAGGAGTTTCTCAA (200 nM) | |||||
| P2 | 213~239 | (ATT)8 | F: AAATCAATAAATATTACGAGTTTTCCA (100 nM) | 8 | MW921470 |
| R: TTTTTATCGTTCAAAATCCTTCTG (200 nM) | |||||
| P3 | 256~264 | (TGA)6 | F: GGGGCCTCCTCATAAGTACC (100 nM) | 5 | MW921471 |
| R: TGCATTTCTTGCACTCCATT (200 nM) | |||||
| P4 | 348~359 | (CAA)6 + (CCA)6 | F: CCCACGCACTAACGAGAGTT (150 nM) | 5 | MW921472 |
| R: AGCGGGAGTTCCATTTCTCT (200 nM) | |||||
| P5 | 453~465 | (TTC)7 | F: TCCTCACCCCAAAAAGTGAC (100 nM) | 6 | MW921473 |
| R: AGGGAGCGACTTCAAATCAA (200 nM) |
| Population | Locus | N | Na | Ne | Ho | He | uHe | F |
|---|---|---|---|---|---|---|---|---|
| TC_DKMZ | P1 | 95 | 4.000 | 2.306 | 0.589 | 0.566 | 0.569 | −0.041 |
| P2 | 95 | 4.000 | 1.137 | 0.126 | 0.120 | 0.121 | −0.048 | |
| P3 | 95 | 2.000 | 1.134 | 0.126 | 0.118 | 0.119 | −0.067 | |
| P4 | 95 | 4.000 | 1.367 | 0.263 | 0.268 | 0.270 | 0.019 | |
| P5 | 95 | 4.000 | 1.149 | 0.116 | 0.130 | 0.130 | 0.107 | |
| CH_HRHT | P1 | 94 | 4.000 | 2.223 | 0.596 | 0.550 | 0.553 | −0.083 |
| P2 | 94 | 5.000 | 1.242 | 0.160 | 0.195 | 0.196 | 0.181 | |
| P3 | 94 | 5.000 | 1.229 | 0.181 | 0.186 | 0.187 | 0.028 | |
| P4 | 94 | 4.000 | 1.570 | 0.383 | 0.363 | 0.365 | −0.055 | |
| P5 | 94 | 4.000 | 1.366 | 0.245 | 0.268 | 0.269 | 0.087 | |
| PT_MJ | P1 | 80 | 3.000 | 1.666 | 0.363 | 0.400 | 0.402 | 0.094 |
| P2 | 81 | 4.000 | 1.190 | 0.173 | 0.160 | 0.161 | −0.082 | |
| P3 | 81 | 2.000 | 1.261 | 0.210 | 0.207 | 0.208 | −0.014 | |
| P4 | 81 | 3.000 | 1.412 | 0.272 | 0.292 | 0.293 | 0.069 | |
| P5 | 81 | 3.000 | 1.191 | 0.173 | 0.160 | 0.161 | −0.077 | |
| KH_BGZY | P1 | 48 | 3.000 | 2.251 | 0.500 | 0.556 | 0.562 | 0.100 |
| P2 | 48 | 5.000 | 1.549 | 0.333 | 0.355 | 0.358 | 0.060 | |
| P3 | 48 | 3.000 | 1.317 | 0.271 | 0.241 | 0.243 | −0.125 | |
| P4 | 48 | 3.000 | 1.539 | 0.250 | 0.350 | 0.354 | 0.286 | |
| P5 | 48 | 4.000 | 1.186 | 0.167 | 0.157 | 0.159 | −0.062 | |
| HL_BAXC | P1 | 47 | 5.000 | 1.958 | 0.511 | 0.489 | 0.495 | −0.043 |
| P2 | 47 | 7.000 | 2.345 | 0.532 | 0.574 | 0.580 | 0.073 | |
| P3 | 47 | 2.000 | 1.394 | 0.213 | 0.282 | 0.286 | 0.247 | |
| P4 | 47 | 5.000 | 1.246 | 0.170 | 0.198 | 0.200 | 0.139 | |
| P5 | 47 | 2.000 | 1.394 | 0.255 | 0.282 | 0.286 | 0.096 | |
| HL_DKXL | P1 | 48 | 6.000 | 2.313 | 0.667 | 0.568 | 0.574 | −0.174 |
| P2 | 48 | 7.000 | 2.956 | 0.688 | 0.662 | 0.669 | −0.039 | |
| P3 | 48 | 2.000 | 1.110 | 0.104 | 0.099 | 0.100 | −0.055 | |
| P4 | 48 | 4.000 | 1.135 | 0.104 | 0.119 | 0.120 | 0.126 | |
| P5 | 48 | 3.000 | 1.567 | 0.333 | 0.362 | 0.366 | 0.079 | |
| HL_GHJA | P1 | 48 | 5.000 | 1.945 | 0.500 | 0.486 | 0.491 | −0.029 |
| P2 | 48 | 7.000 | 2.855 | 0.646 | 0.650 | 0.657 | 0.006 | |
| P3 | 48 | 2.000 | 1.087 | 0.083 | 0.080 | 0.081 | −0.043 | |
| P4 | 48 | 3.000 | 1.290 | 0.208 | 0.225 | 0.227 | 0.072 | |
| P5 | 48 | 3.000 | 1.640 | 0.333 | 0.390 | 0.395 | 0.146 | |
| YL_NFSA | P1 | 47 | 5.000 | 2.392 | 0.574 | 0.582 | 0.588 | 0.013 |
| P2 | 47 | 6.000 | 2.295 | 0.596 | 0.564 | 0.570 | −0.056 | |
| P3 | 47 | 2.000 | 1.160 | 0.106 | 0.138 | 0.139 | 0.228 | |
| P4 | 47 | 4.000 | 1.508 | 0.298 | 0.337 | 0.341 | 0.116 | |
| P5 | 47 | 3.000 | 1.655 | 0.447 | 0.396 | 0.400 | −0.129 | |
| HL_RRRS | P1 | 47 | 5.000 | 2.179 | 0.638 | 0.541 | 0.547 | −0.180 |
| P2 | 46 | 6.000 | 2.635 | 0.587 | 0.621 | 0.627 | 0.054 | |
| P3 | 47 | 2.000 | 1.209 | 0.106 | 0.173 | 0.175 | 0.386 | |
| P4 | 47 | 4.000 | 1.462 | 0.213 | 0.316 | 0.319 | 0.327 | |
| P5 | 47 | 2.000 | 1.367 | 0.277 | 0.268 | 0.271 | −0.031 | |
| HL_TCGF | P1 | 48 | 5.000 | 2.383 | 0.583 | 0.580 | 0.586 | −0.005 |
| P2 | 48 | 7.000 | 3.130 | 0.708 | 0.681 | 0.688 | −0.041 | |
| P3 | 48 | 3.000 | 1.158 | 0.104 | 0.137 | 0.138 | 0.237 | |
| P4 | 48 | 4.000 | 1.383 | 0.271 | 0.277 | 0.280 | 0.023 | |
| P5 | 48 | 2.000 | 1.358 | 0.271 | 0.264 | 0.266 | −0.027 | |
| HL_ZHFL | P1 | 48 | 6.000 | 1.801 | 0.458 | 0.445 | 0.450 | −0.030 |
| P2 | 48 | 6.000 | 2.122 | 0.625 | 0.529 | 0.534 | −0.182 | |
| P3 | 48 | 2.000 | 1.180 | 0.167 | 0.153 | 0.154 | −0.091 | |
| P4 | 48 | 3.000 | 1.135 | 0.125 | 0.119 | 0.120 | −0.051 | |
| P5 | 48 | 2.000 | 1.679 | 0.354 | 0.404 | 0.409 | 0.124 |
| TC_DKMZ | CH_HRHT | PT_MJ | KH_BGZY | HL_BAXC | HL_DKXL | HL_GHJA | YL_NFSA | HL_RRRS | HL_TCGF | HL_ZHFL | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TC_DKMZ | 0.000 | 0.014 | 0.004 | 0.020 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 |
| CH_HRHT | 0.009 | 0.000 | 0.001 | 0.014 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 |
| PT_MJ | 0.017 | 0.026 | 0.000 | 0.014 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 |
| KH_BGZY | 0.012 | 0.014 | 0.014 | 0.000 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 |
| HL_BAXC | 0.073 | 0.057 | 0.062 | 0.034 | 0.000 | 0.065 | 0.090 | 0.028 | 0.105 | 0.068 | 0.109 |
| HL_DKXL | 0.106 | 0.083 | 0.103 | 0.058 | 0.008 | 0.000 | 0.437 | 0.062 | 0.053 | 0.295 | 0.196 |
| HL_GHJA | 0.101 | 0.074 | 0.089 | 0.053 | 0.007 | 0.000 | 0.000 | 0.141 | 0.103 | 0.179 | 0.396 |
| YL_NFSA | 0.068 | 0.043 | 0.072 | 0.028 | 0.011 | 0.010 | 0.005 | 0.000 | 0.003 | 0.011 | 0.021 |
| HL_RRRS | 0.105 | 0.086 | 0.091 | 0.052 | 0.006 | 0.009 | 0.006 | 0.020 | 0.000 | 0.413 | 0.018 |
| HL_TCGF | 0.100 | 0.084 | 0.095 | 0.044 | 0.009 | 0.002 | 0.004 | 0.014 | 0.000 | 0.000 | 0.010 |
| HL_ZHFL | 0.110 | 0.084 | 0.095 | 0.066 | 0.007 | 0.004 | 0.000 | 0.016 | 0.015 | 0.018 | 0.000 |
| TC_DKMZ | CH_HRHT | PT_MJ | KH_BGZY | HL_BAXC | HL_DKXL | HL_GHJA | YL_NFSA | HL_RRRS | HL_TCGF | HL_ZHFL | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TC_DKMZ | 0.000 | ||||||||||
| CH_HRHT | 27.571 | 0.000 | |||||||||
| PT_MJ | 14.584 | 9.387 | 0.000 | ||||||||
| KH_BGZY | 19.947 | 18.185 | 17.746 | 0.000 | |||||||
| HL_BAXC | 3.186 | 4.099 | 3.780 | 7.172 | 0.000 | ||||||
| HL_DKXL | 2.108 | 2.762 | 2.166 | 4.039 | 29.808 | 0.000 | |||||
| HL_GHJA | 2.218 | 3.124 | 2.565 | 4.459 | 36.629 | n.a. | 0.000 | ||||
| YL_NFSA | 3.444 | 5.517 | 3.246 | 8.617 | 21.595 | 25.792 | 48.282 | 0.000 | |||
| HL_RRRS | 2.129 | 2.642 | 2.502 | 4.574 | 38.291 | 27.387 | 38.790 | 12.212 | 0.000 | ||
| HL_TCGF | 2.254 | 2.711 | 2.383 | 5.370 | 28.100 | 119.469 | 61.551 | 18.122 | n.a. | 0.000 | |
| HL_ZHFL | 2.027 | 2.717 | 2.384 | 3.520 | 37.650 | 63.288 | n.a. | 15.118 | 16.698 | 13.920 | 0.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ching, Y.-H.; Kuo, Y.-C.; Su, M.-C.; Wang, S.-C.; Lin, C.-F.; Tu, W.-C.; Lin, M.-D. Genetic Differentiation of the Bloodsucking Midge Forcipomyia taiwana (Diptera: Ceratopogonidae): Implication of the Geographic Isolation by the Central Mountain Ranges in Taiwan. Insects 2024, 15, 23. https://doi.org/10.3390/insects15010023
Ching Y-H, Kuo Y-C, Su M-C, Wang S-C, Lin C-F, Tu W-C, Lin M-D. Genetic Differentiation of the Bloodsucking Midge Forcipomyia taiwana (Diptera: Ceratopogonidae): Implication of the Geographic Isolation by the Central Mountain Ranges in Taiwan. Insects. 2024; 15(1):23. https://doi.org/10.3390/insects15010023
Chicago/Turabian StyleChing, Yung-Hao, Yuan-Chen Kuo, Ming-Ching Su, Szu-Chieh Wang, Chuen-Fu Lin, Wu-Chun Tu, and Ming-Der Lin. 2024. "Genetic Differentiation of the Bloodsucking Midge Forcipomyia taiwana (Diptera: Ceratopogonidae): Implication of the Geographic Isolation by the Central Mountain Ranges in Taiwan" Insects 15, no. 1: 23. https://doi.org/10.3390/insects15010023