
Combined PacBio Iso-Seq and Illumina RNA-Seq Analysis of the Tuta absoluta (Meyrick) Transcriptome and Cytochrome P450 Genes
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Insect Culture
2.2. Sample Processing
2.3. RNA Isolation and Sequencing
2.4. Statistical Analyses
2.4.1. Error Correction and Analysis of PacBio Iso-Seq Data
2.4.2. RNA-Seq Analysis of T. absoluta
2.4.3. Functional Annotation and Enrichment Analysis of DETs
2.4.4. Analysis of the P450 Gene
2.4.5. RNAseq Validation by Quantitative PCR
3. Results
3.1. Summary of RNA-Seq and SMRT Sequencing Results
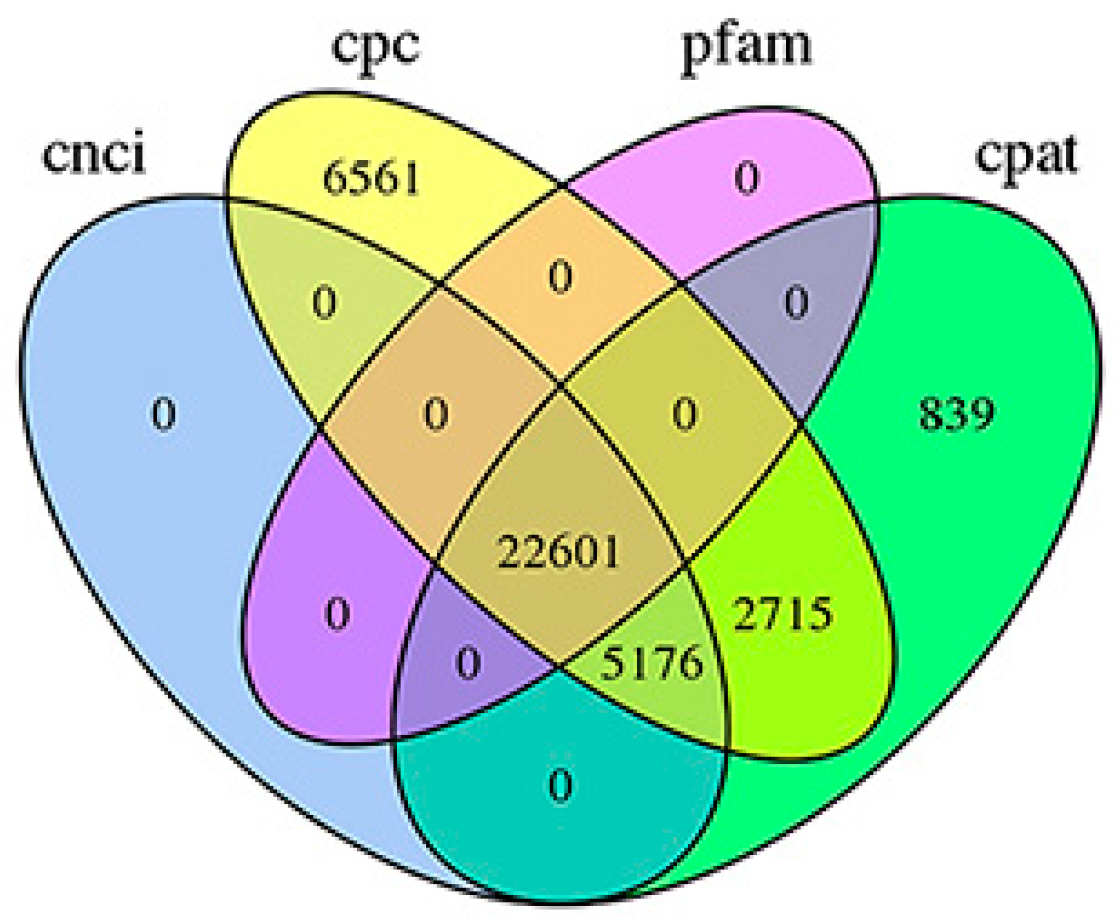
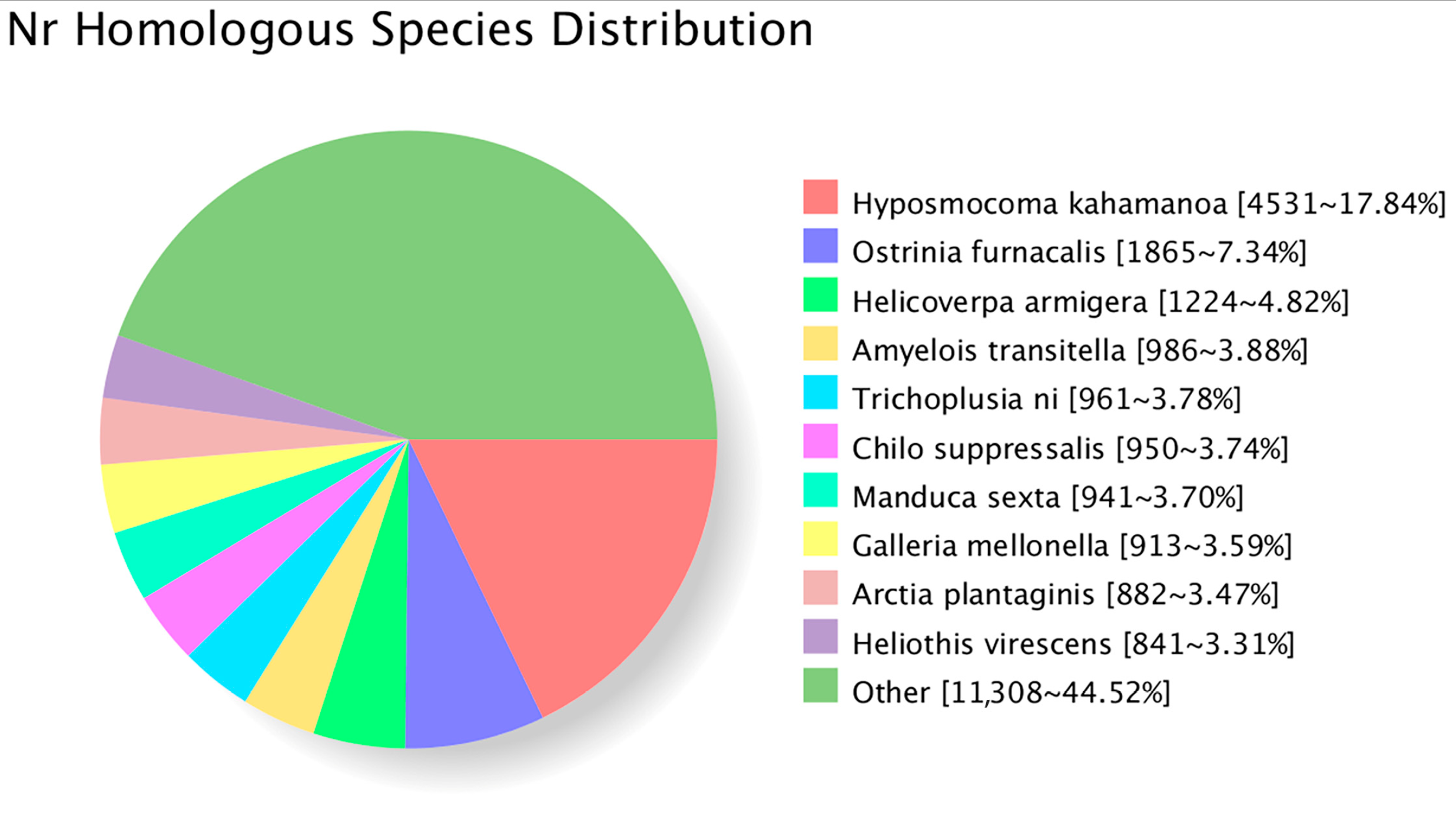
3.2. Structural Analysis
3.3. Quantitative Analysis of Transcript Expression
3.4. DETs Analysis
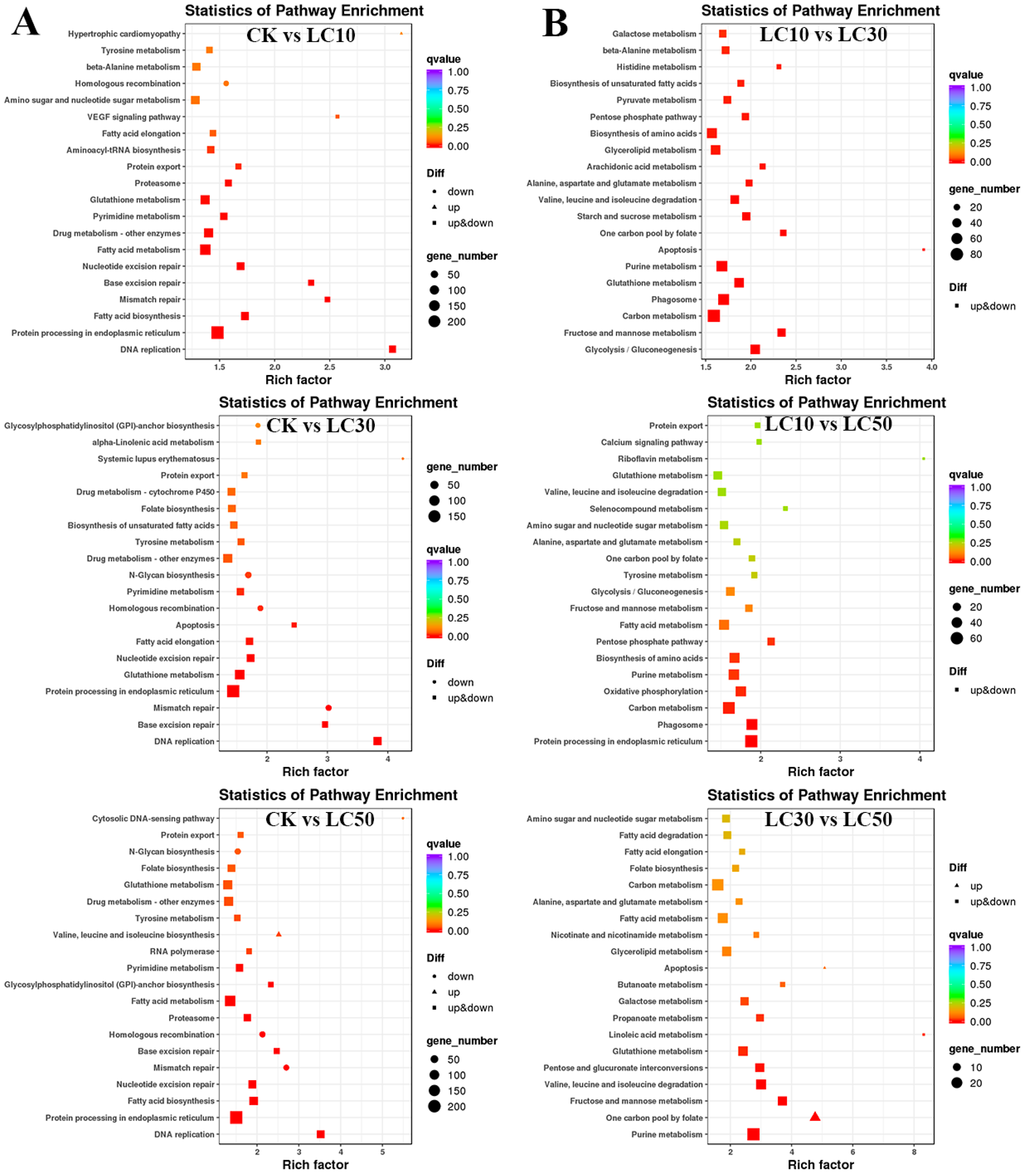
3.5. Functional Annotation and Enrichment Analysis of DETs
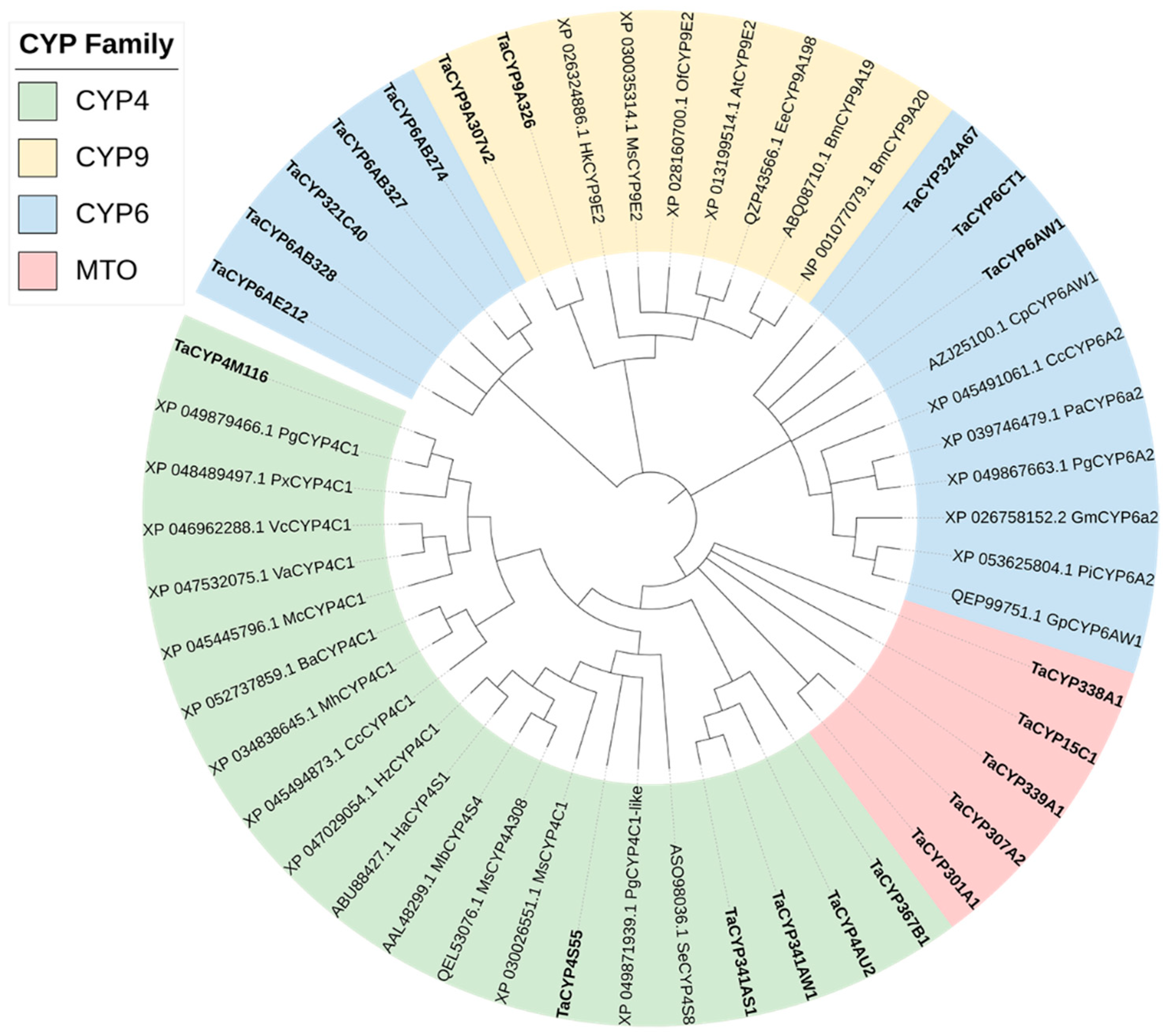
3.6. CYP Analysis
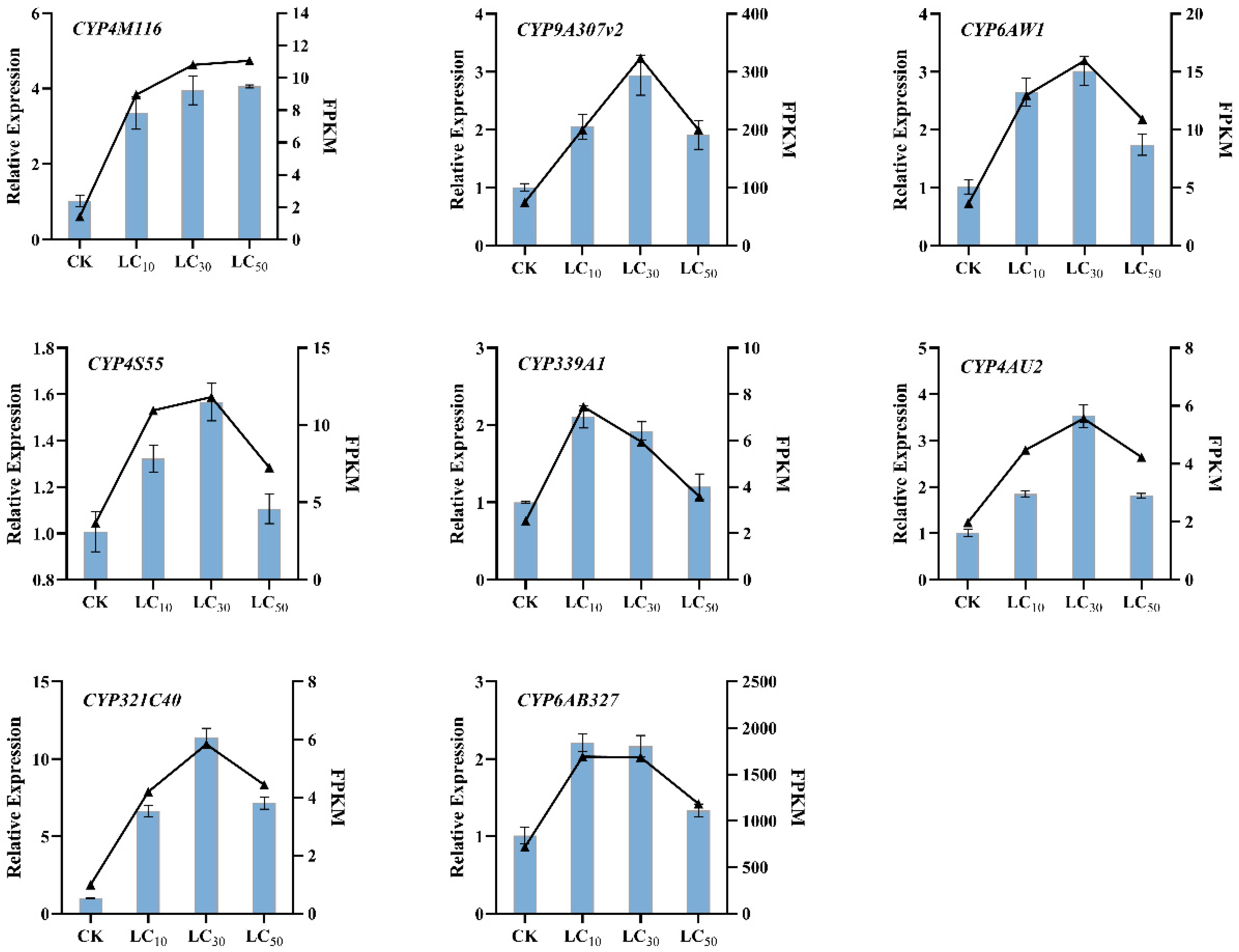
3.7. RNAseq Validation by Quantitative PCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Biondi, A.; Guedes, R.N.; Wan, F.H.; Desneux, N. Ecology, Worldwide Spread, and Management of the Invasive South American Tomato Pinworm, Tuta absoluta: Past, Present, and Future. Annu. Rev. Entomol. 2018, 63, 239–258. [Google Scholar] [CrossRef]
- Desneux, N.; Wajnberg, E.; Wyckhuys, K.; Burgio, G.; Arpaia, S.; Narváez-Vasquez, C.; González-Cabrera, J.; Ruescas, D.C.; Tabone, E.; Frandon, J. Biological invasion of European tomato crops by Tuta absoluta: Ecology, geographic expansion and prospects for biological control. J. Pest Sci. 2010, 83, 197–215. [Google Scholar] [CrossRef]
- Guimapi, R.A.; Srinivasan, R.; Tonnang, H.E.; Sotelo-Cardona, P.; Mohamed, S.A. Exploring the Mechanisms of the Spatiotemporal Invasion of Tuta absoluta in Asia. Agriculture 2020, 10, 124. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.-F.; Wang, Y.-S.; Gao, Y.-H.; Liu, W.-X.; Zhang, R.; Fu, W.-J.; Xian, X.-Q.; Jun, W.; Kuang, M.; Wan, F.-H. First report of the South American tomato leafminer, Tuta absoluta (Meyrick), in China. J. Integr. Agric. 2020, 19, 1912–1917. [Google Scholar] [CrossRef]
- Xian, X.; Han, P.; Wang, S.; Zhang, G.; Liu, W.; Desneux, N.; Wan, F. The potential invasion risk and preventive measures against the tomato leafminer Tuta absoluta in China. Entomol. Gen. 2017, 36, 319–333. [Google Scholar] [CrossRef]
- Zhang, G.-F.; Xian, X.-Q.; Zhang, Y.-B.; Liu, W.-X.; Liu, H.; Feng, X.-D.; Ma, D.-Y.; Wang, Y.-S.; Gao, Y.-H.; Zhang, R.; et al. Outbreak of the South American tomato leafminer, Tuta absoluta, in the Chinese mainland: Geographic and potential host range expansion. Pest Manag. Sci. 2021, 77, 5475–5488. [Google Scholar] [CrossRef]
- Zhang, G.F.; Ma, D.Y.; Liu, W.X.; Wang, Y.S.; Fu, W.J.; Wang, J.; Gao, Y.H.; Wang, F.H. A newly discovered invasive pest in China-Tuta absoluta (Lepidoptera: Gelechiidae). J. Biosafety 2019, 28, 200–203. [Google Scholar]
- Guedes, R.N.C.; Roditakis, E.; Campos, M.R.; Haddi, K.; Bielza, P.; Siqueira, H.A.A.; Tsagkarakou, A.; Vontas, J.; Nauen, R. Insecticide resistance in the tomato pinworm Tuta absoluta: Patterns, spread, mechanisms, management and outlook. J. Pest Sci. 2019, 92, 1329–1342. [Google Scholar] [CrossRef]
- Silva, J.E.; Ribeiro, L.M.D.; Vinasco, N.; Guedes, R.N.C.; Siqueira, H.A.A. Field-evolved resistance to chlorantraniliprole in the tomato pinworm Tuta absoluta: Inheritance, cross-resistance profile, and metabolism. J. Pest Sci. 2019, 92, 1421–1431. [Google Scholar] [CrossRef]
- Cherif, A.; Harbaoui, K.; Zappalà, L.; Grissa-Lebdi, K. Efficacy of mass trapping and insecticides to control Tuta absoluta in Tunisia. J. Plant Dis. Prot. 2017, 125, 51–61. [Google Scholar] [CrossRef]
- Yang, L.; Xing, B.; Li, F.; Wang, L.K.; Yuan, L.; Mbuji, A.L.; Peng, Z.; Malhat, F.; Wu, S. Full-length transcriptome analysis of Spodoptera frugiperda larval brain reveals detoxification genes. Peerj 2021, 9, e12069. [Google Scholar] [CrossRef] [PubMed]
- Pavek, P.; Dvorak, Z. Xenobiotic-Induced Transcriptional Regulation of Xenobiotic Metabolizing Enzymes of the Cytochrome P450 Superfamily in Human Extrahepatic Tissues. Curr. Drug Metab. 2008, 9, 129–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feyereisen, R. Arthropod CYPomes illustrate the tempo and mode in P450 evolution. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2011, 1814, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, M.; Wang, Y.; Song, W.; Tang, P. Identification and functional analysis of cytochrome P450 CYP346 family genes associated with phosphine resistance in Tribolium castaneum. Pestic. Biochem. Physiol. 2020, 168, 104622. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the potential of full-length transcriptome sequencing. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20190097. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M. Widespread Polycistronic Transcripts in Fungi Revealed by Single-Molecule mRNA Sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.; Underwood, J.G.; Tseng, E.; Holloway, A.K.; Bench Basinet Cv, D.C.I. Long-Read Sequencing of Chicken Transcripts and Identification of New Transcript Isoforms. PLoS ONE 2014, 9, e94650. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Yang, P.; Xie, S.; Shi, M.; Huang, J.; Wang, Z.; Chen, X. Comparative Transcriptome Analysis Reveals Sex-Based Differences during the Development of the Adult Parasitic Wasp Cotesia vestalis (Hymenoptera: Braconidae). Genes 2021, 12, 896. [Google Scholar] [CrossRef]
- Yang, H.; Xu, D.; Zhuo, Z.; Hu, J.; Lu, B. SMRT sequencing of the full-length transcriptome of the Rhynchophorus ferrugineus (Coleoptera: Curculionida). Peerj 2020, 8, e91333. [Google Scholar] [CrossRef]
- Xu, D.; Yang, H.; Zhuo, Z.; Lu, B.; Hu, J.; Yang, F. Characterization and analysis of the transcriptome in Opisina arenosella from different developmental stages using single-molecule real-time transcript sequencing and RNA-seq. Int. J. Biol. Macromol. 2021, 169, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Mei, W.; Soltis, P.S.; Soltis, D.E.; Barbazuk, W.B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol. Ecol. Resour. 2017, 17, 1243–1256. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Yeh, L. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Jianqi, L.I.; Songfeng, W.U.; Zhu, Y.; Chen, Y.; Fuchu, H.E. Integrated nr Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Cherry, J.M. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Alex, B.; Jody, C.; Penelope, C.; Eberhardt, R.Y.; Eddy, S.R.; Andreas, H.; Kirstie, H.; Liisa, H.; Jaina, M. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Noriega, D.D.; Arias, P.L.; Barbosa, H.R.; Arraes, F.B.M.; Ossa, G.A.; Villegas, B.; Coelho, R.R.; Albuquerque, E.V.S.; Togawa, R.C.; Grynberg, P.; et al. Transcriptome and gene expression analysis of three developmental stages of the coffee berry borer, Hypothenemus hampei. Sci. Rep. 2019, 9, 12804. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, X.Q.; Zhang, L. Identification of putative odorant binding proteins in the peach fruit borer Carposina sasakii Matsumura (Lepidoptera: Carposinidae) by transcriptome analysis and their expression profile. Biochem. Biophys. Res. Commun. 2019, 508, 1024–1030. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, X.; Lei, C.; Zhu, F. Sensory genes identification with head transcriptome of the migratory armyworm, Mythimna separata. Sci. Rep. 2017, 7, 46033. [Google Scholar] [CrossRef] [Green Version]
- Fu, G.; Condon, K.C.; Epton, M.J.; Gong, P.; Jin, L.; Condon, G.C.; Morrison, N.I.; Dafa’alla, T.H.; Alphey, L. Female-specific insect lethality engineered using alternative splicing. Nat. Biotechnol. 2007, 25, 353–357. [Google Scholar] [CrossRef]
- Fahmi, N.A.; Nassereddeen, H.; Chang, J.; Park, M.; Yeh, H.; Sun, J.; Fan, D.; Yong, J.; Zhang, W. AS-Quant: Detection and Visualization of Alternative Splicing Events with RNA-seq Data. Int. J. Mol. Sci. 2021, 22, 4468. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Huang, B.; Xu, Y.M.; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H.; Yang, W.M.; et al. Mechanism of alternative splicing and its regulation (Review). Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Tilgner, H.; Grubert, F.; Sharon, D.; Snyder, M.P. Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proc. Natl. Acad. Sci. USA 2014, 111, 9869–9874. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Sharma, S.; Meghwanshi, K.K.; Patel, S.; Mehta, P.; Shukla, N.; Do, D.N.; Rajpurohit, S.; Suravajhala, P.; Shukla, J.N. Long Non-Coding RNAs in Insects. Animals 2021, 11, 1118. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.B.; Boley, N.; Eisman, R.; May, G.E.; Stoiber, M.H.; Duff, M.O.; Booth, B.W.; Wen, J.; Park, S.; Suzuki, A.M.; et al. Diversity and dynamics of the Drosophila transcriptome. Nature 2014, 512, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graveley, B.R.; Brooks, A.N.; Carlson, J.W.; Duff, M.O.; Landolin, J.M.; Yang, L.; Artieri, C.G.; van Baren, M.J.; Boley, N.; Booth, B.W.; et al. The developmental transcriptome of Drosophila melanogaster. Nature 2011, 471, 473–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soshnev, A.A.; Ishimoto, H.; McAllister, B.F.; Li, X.; Wehling, M.D.; Kitamoto, T.; Geyer, P.K. A Conserved Long Noncoding RNA Affects Sleep Behavior in Drosophila. Genetics 2011, 189, 455–468. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.W.; Yuan, G.R.; Chen, M.L.; Dou, W.; Jing, T.X.; Zheng, L.S.; Peng, M.L.; Bai, W.J.; Wang, J.J. Genome-wide identification of long non-coding RNAs (lncRNAs) associated with malathion resistance in Bactrocera dorsalis. Pest Manag. Sci. 2021, 77, 2292–2301. [Google Scholar] [CrossRef]
- Qiao, H.L.; Wang, J.Y.; Wang, Y.Z.; Yang, J.J.; Wei, B.F.; Li, M.M.; Wang, B.; Li, X.Z.; Cao, Y.; Tian, L.; et al. Transcriptome analysis reveals potential function of long non-coding RNAs in 20-hydroxyecdysone regulated autophagy in Bombyx mori. BMC Genom. 2021, 22, 374. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, T.; He, W.; Shen, X.; Zhao, Q.; Bai, J.; You, M. Genome-wide identification and characterization of putative lncRNAs in the diamondback moth, Plutella xylostella (L.). Genomics 2018, 110, 35–42. [Google Scholar] [CrossRef]
- Li, W.-J.; Song, Y.-J.; Han, H.-L.; Xu, H.-Q.; Wei, D.; Smagghe, G.; Wang, J.-J. Genome-wide analysis of long non-coding RNAs in adult tissues of the melon fly, Zeugodacus cucurbitae (Coquillett). BMC Genom. 2020, 21, 600. [Google Scholar] [CrossRef] [PubMed]
- Kodrik, D.; Bednarova, A.; Zemanova, M.; Krishnan, N. Hormonal Regulation of Response to Oxidative Stress in Insects-An Update. Int. J. Mol. Sci. 2015, 16, 25788–25816. [Google Scholar] [CrossRef] [Green Version]
- Kodrik, D.; Krishnan, N.; Habustova, O. Is the titer of adipokinetic peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by oxidative stress? Peptides 2007, 28, 974–980. [Google Scholar] [CrossRef]
- Schuler, M.A. P450s in plant-insect interactions. Biochim. Biophys. Acta-Proteins Proteom. 2011, 1814, 36–45. [Google Scholar] [CrossRef]
- Lu, K.; Song, Y.; Zeng, R. The role of cytochrome P450-mediated detoxification in insect adaptation to xenobiotics. Curr. Opin. Insect Sci. 2021, 43, 103–107. [Google Scholar] [CrossRef]
- Stanley-Samuelson, D.W.; Jurenka, R.A.; Cripps, C.; Blomquist, G.J.; de Renobales, M. Fatty acids in insects: Composition, metabolism, and biological significance. Arch. Insect Biochem. Physiol. 1988, 9, 1–33. [Google Scholar] [CrossRef]
- Huang, Z.; Zhao, M.; Shi, P. Sublethal effects of azadirachtin on lipid metabolism and sex pheromone biosynthesis of the Asian corn borer Ostrinia furnacalis. Phytoparasitica 2012, 40, 361–368. [Google Scholar] [CrossRef]
- Guo, Y.; Chai, Y.; Zhang, L.; Zhao, Z.; Gao, L.-L.; Ma, R. Transcriptome Analysis and Identification of Major Detoxification Gene Families and Insecticide Targets in Grapholita molesta (Busck) (Lepidoptera: Tortricidae). J. Insect Sci. 2017, 17, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Nardini, L.; Christian, R.N.; Coetzer, N.; Ranson, H.; Coetzee, M.; Koekemoer, L.L. Detoxification enzymes associated with insecticide resistance in laboratory strains of Anopheles arabiensis of different geographic origin. Parasites Vectors 2012, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Schuler, M.A.; Berenbaum, M.R. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu. Rev. Entomol. 2007, 52, 231–253. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Chen, F.; Yu, S.J.; Ding, L.L.; Yang, J.; Luo, R.; Tian, H.X.; Li, H.J.; Liu, H.Q.; Ran, C. Transcriptome and Difference Analysis of Fenpropathrin Resistant Predatory Mite, Neoseiulus barkeri (Hughes). Int. J. Mol. Sci. 2016, 17, 704. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.J.; Zhang, Y.H.; Li, J.; Fang, J.P.; Liu, T.T.; Dong, L.Y. Transcriptome profiling to identify cytochrome P450 genes involved in penoxsulam resistance in Echinochloa glabrescens. Pestic. Biochem. Physiol. 2019, 158, 112–120. [Google Scholar] [CrossRef]
- Wan, L.R.; Zhou, A.L.; Xiao, W.F.; Zou, B.X.; Jiang, Y.M.; Xiao, J.S.; Deng, C.; Zhang, Y.H.; Huang, Z.Y.; Bu, C.F.; et al. Cytochrome P450 monooxygenase genes in the wild silkworm, Bombyx mandarina. Peerj 2021, 9, e10818. [Google Scholar] [CrossRef]
- Stavrakaki, M.; Ilias, A.; Ioannidis, P.; Vontas, J.; Roditakis, E. Investigating mechanisms associated with emamectin benzoate resistance in the tomato borer Tuta absoluta. J. Pest Sci. 2022, 95, 1163–1177. [Google Scholar] [CrossRef]
- Denholm, I.; Pickett, J.A.; Devonshire, A.L.; Bergé, J.; Feyereisen, R.; Amichot, M. Cytochrome P450 monooxygenases and insecticide resistance in insects. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 1998, 353, 1701–1705. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, X.; Bai, R.; Shi, Y.; Tang, Q.; An, S.; Song, Q.; Yan, F. RNA interference of the P450 CYP6CM1 gene has different efficacy in B and Q biotypes of Bemisia tabaci. Pest Manag. Sci. 2015, 71, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, J.; Wu, H.; Zhang, H.; Zhang, J.; Ma, E. Knockdown of cytochrome P450 CYP6 family genes increases susceptibility to carbamates and pyrethroids in the migratory locust, Locusta migratoria. Chemosphere 2019, 223, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.; Williams, D.R.; Aguiar-Santana, I.A.; Pedersen, J.; Turner, P.C.; Rees, H.H. Expression and down-regulation of cytochrome P450 genes of the CYP4 family by ecdysteroid agonists in Spodoptera littoralis and Drosophila melanogaster. Insect Biochem. Mol. Biol. 2006, 36, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Ranson, H.; Nikou, D.; Hutchinson, M.; Wang, X.; Roth, C.W.; Hemingway, J.; Collins, F.H. Molecular analysis of multiple cytochrome P450 genes from the malaria vector, Anopheles gambiae. Insect Mol. Biol. 2002, 11, 409–418. [Google Scholar] [CrossRef]
- Zhou, X.; Sheng, C.; Li, M.; Wan, H.; Liu, D.; Qiu, X. Expression responses of nine cytochrome P450 genes to xenobiotics in the cotton bollworm Helicoverpa armigera. Pestic. Biochem. Physiol. 2010, 97, 209–213. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Total Number of Pair-End Reads | Base Number | GC (%) | Q20 (%) | Q30 (%) | Total Mapped (%) |
|---|---|---|---|---|---|---|
| CK1 | 29,371,782 | 8,766,740,538 | 48.28 | 98.32 | 95.46 | 90.96 |
| CK2 | 25,534,773 | 7,623,374,636 | 48.70 | 98.23 | 95.23 | 91.40 |
| CK3 | 20,126,926 | 6,014,056,388 | 48.63 | 98.23 | 95.16 | 89.89 |
| LC10-1 | 21,785,788 | 6,500,875,664 | 49.22 | 97.60 | 93.69 | 89.60 |
| LC10-2 | 24,525,334 | 7,315,451,512 | 49.14 | 97.71 | 93.96 | 89.28 |
| LC10-3 | 26,961,547 | 8,041,765,962 | 49.06 | 97.42 | 93.39 | 88.77 |
| LC30-1 | 26,889,981 | 8,022,376,368 | 49.69 | 97.51 | 93.57 | 89.55 |
| LC30-2 | 26,987,086 | 8,045,001,412 | 49.43 | 97.39 | 93.34 | 88.60 |
| LC30-3 | 24,346,249 | 7,267,996,486 | 49.40 | 97.43 | 93.41 | 89.13 |
| LC50-1 | 26,029,165 | 7,771,009,310 | 49.82 | 97.18 | 92.87 | 90.12 |
| LC50-2 | 31,441,769 | 9,386,493,344 | 49.59 | 97.88 | 94.15 | 90.83 |
| LC50-3 | 30,015,728 | 8,957,881,670 | 49.44 | 98.21 | 95.23 | 90.38 |
| DET Set | All DET | Upregulated | Downregulated |
|---|---|---|---|
| CK vs. LC10 | 9414 | 4146 | 5268 |
| CK vs. LC30 | 6892 | 3027 | 3865 |
| CK vs. LC50 | 8162 | 3581 | 4581 |
| LC10 vs. LC30 | 2583 | 1609 | 974 |
| LC10 vs. LC50 | 1848 | 1054 | 794 |
| LC30 vs. LC50 | 669 | 356 | 313 |
| DET Set | Total | COG | GO | KEGG | KOG | NR | Pfam | Swiss-Prot | TrEMBL | eggNOG |
|---|---|---|---|---|---|---|---|---|---|---|
| CK vs. LC10 | 8711 | 2968 | 7361 | 7081 | 6283 | 8589 | 7236 | 5196 | 8576 | 7306 |
| CK vs. LC30 | 6417 | 2197 | 5403 | 5259 | 4728 | 6339 | 5396 | 3933 | 6330 | 5378 |
| CK vs. LC50 | 7544 | 2492 | 6392 | 6118 | 5520 | 7439 | 6328 | 4510 | 7426 | 6328 |
| LC10 vs.LC30 | 2383 | 879 | 2052 | 1995 | 1779 | 2358 | 2016 | 1564 | 2361 | 2050 |
| LC10 vs.LC50 | 1693 | 626 | 1421 | 1357 | 1198 | 1666 | 1404 | 1074 | 1665 | 1420 |
| LC30 vs.LC50 | 618 | 244 | 531 | 524 | 467 | 610 | 511 | 425 | 611 | 521 |
| Gene ID | Gene Name | Log2FC (CKvsLC10) | Regulated |
|---|---|---|---|
| BMK_Unigene_050674 | CYP367B1 | 1.420509266 | up |
| BMK_Unigene_050670 | CYP4M116 | 2.035326565 | up |
| BMK_Unigene_072885 | CYP341AW1 | 1.022167794 | up |
| BMK_Unigene_051429 | CYP6AW1 | 1.689245983 | up |
| BMK_Unigene_001243 | CYP6AB328 | −1.795651346 | down |
| BMK_Unigene_048006 | CYP339A1 | 1.429081153 | up |
| BMK_Unigene_005085 | CYP9A326 | −1.229421385 | down |
| BMK_Unigene_001201 | CYP9A307v2 | 1.269973516 | up |
| BMK_Unigene_048337 | CYP6AE212 | −1.103023119 | down |
| BMK_Unigene_073841 | CYP4S55 | 1.425289144 | up |
| BMK_Unigene_052034 | CYP307A2 | −1.497466918 | down |
| BMK_Unigene_052058 | CYP15C1 | 1.28923048 | up |
| BMK_Unigene_070417 | CYP338A1 | −3.524270134 | down |
| BMK_Unigene_073442 | CYP321C40 | 1.82095051 | up |
| BMK_Unigene_071946 | CYP301A1 | −1.421794225 | down |
| BMK_Unigene_096077 | CYP6AB327 | 1.106535669 | up |
| BMK_Unigene_093562 | CYP6AB274 | −4.027739403 | down |
| BMK_Unigene_020691 | CYP4AU2 | 1.013224445 | up |
| BMK_Unigene_094349 | CYP6CT1 | −3.306571743 | down |
| BMK_Unigene_096172 | CYP341AS1 | −1.916819856 | down |
| BMK_Unigene_096856 | CYP324A67 | −1.304952081 | down |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Xiao, F.; Zhu, J.; Fu, D.; Wang, Z.; Xiao, R. Combined PacBio Iso-Seq and Illumina RNA-Seq Analysis of the Tuta absoluta (Meyrick) Transcriptome and Cytochrome P450 Genes. Insects 2023, 14, 363. https://doi.org/10.3390/insects14040363
Liu M, Xiao F, Zhu J, Fu D, Wang Z, Xiao R. Combined PacBio Iso-Seq and Illumina RNA-Seq Analysis of the Tuta absoluta (Meyrick) Transcriptome and Cytochrome P450 Genes. Insects. 2023; 14(4):363. https://doi.org/10.3390/insects14040363
Chicago/Turabian StyleLiu, Min, Feng Xiao, Jiayun Zhu, Di Fu, Zonglin Wang, and Rong Xiao. 2023. "Combined PacBio Iso-Seq and Illumina RNA-Seq Analysis of the Tuta absoluta (Meyrick) Transcriptome and Cytochrome P450 Genes" Insects 14, no. 4: 363. https://doi.org/10.3390/insects14040363