
Comparative Mitogenomics of Flesh Flies: Implications for Phylogeny

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and Taxa Identification
2.2. DNA Extraction, Sequencing and Assembly
2.3. Mitogenome Annotation and Sequence Analyses
2.4. Phylogenetic Analysis
3. Results and Discussion
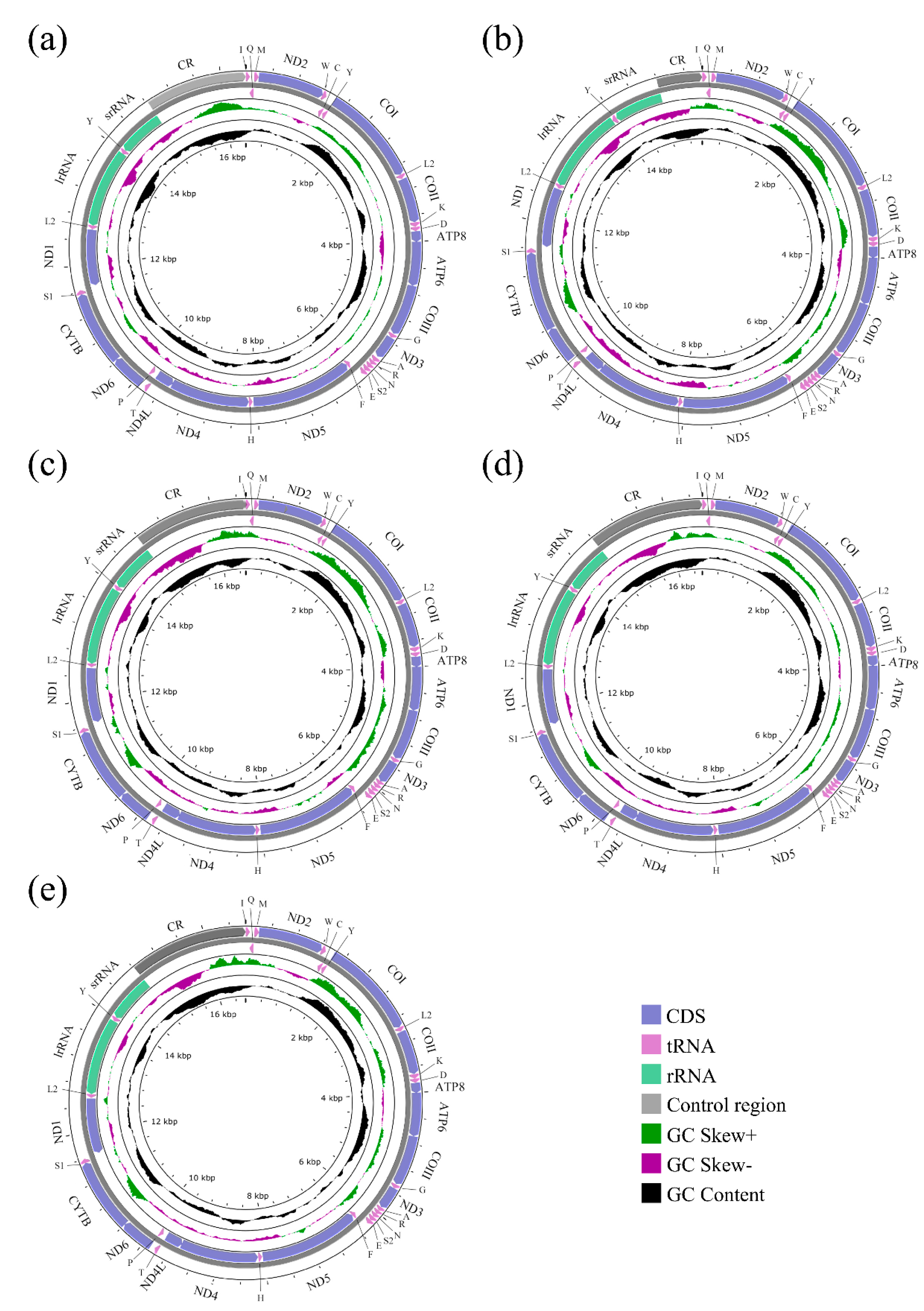
3.1. General Features of Sarcophagid Mitogenomes
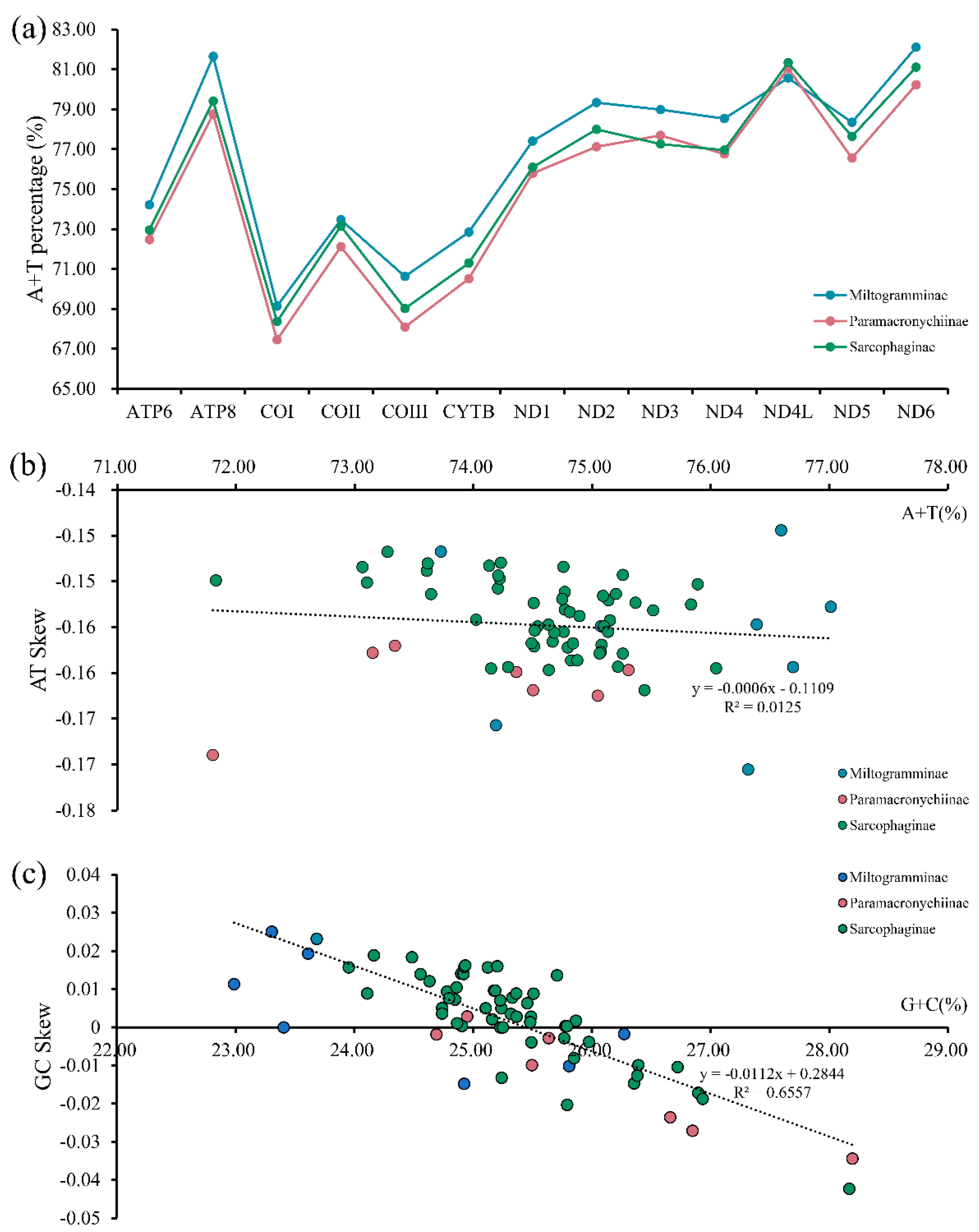
3.2. Protein-Coding Genes and Codon Usage
3.3. Ribosomal, Transfer RNA Genes, and Control Region
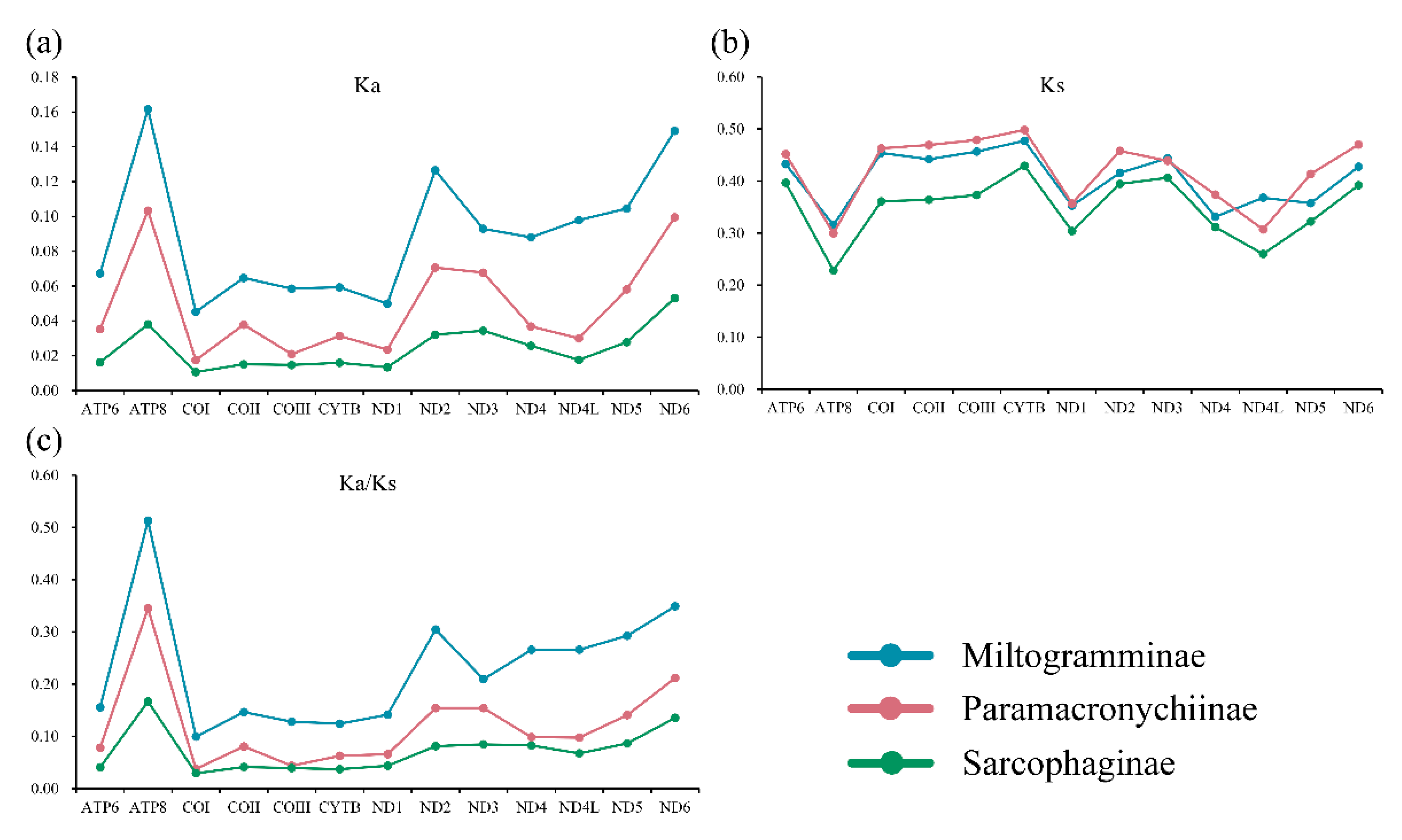
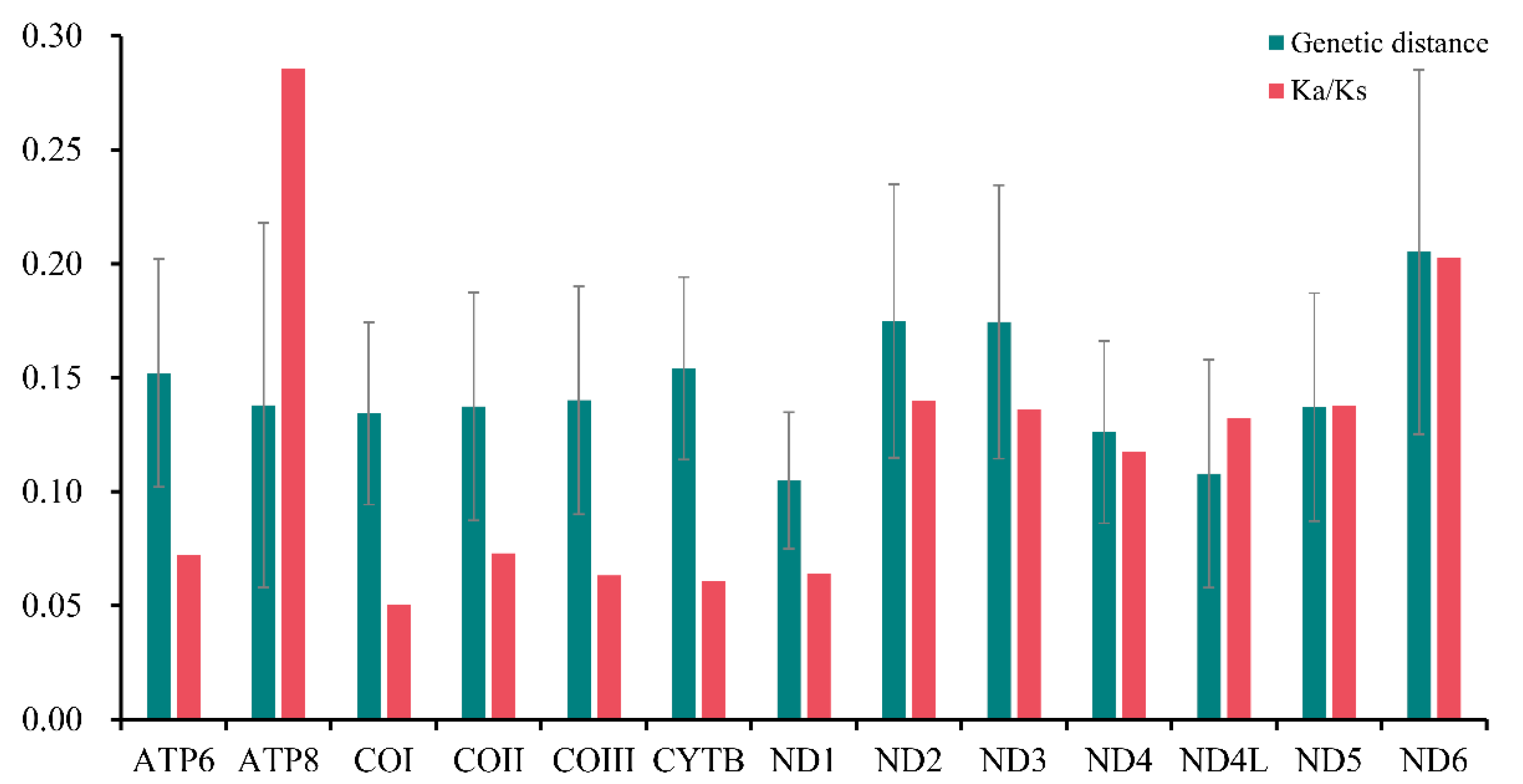
3.4. Evolutionary Rates and Heterogeneous Sequence Divergence
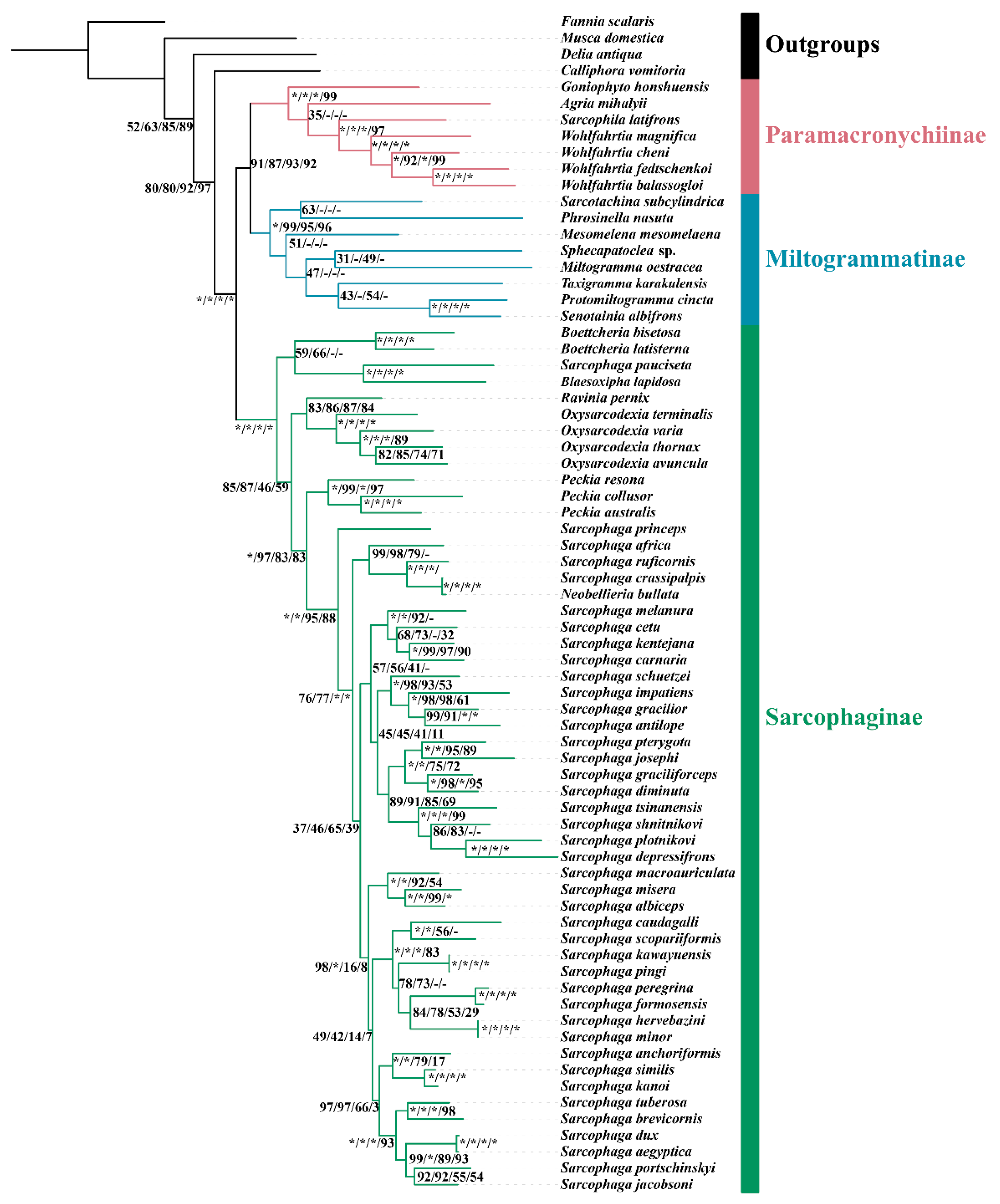
3.5. Phylogenetic Reconstruction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pape, T.; Blagoderov, V.; Mostovski, M.B. Order Diptera Linnaeus, 1758. In Animal Biodiversity: An Outline of Higher-Level Classification and Survey of Taxonomic Richness; Zhang, Z.-Q., Ed.; Magnolia Press: Auckland, New Zealand, 2011; Volume 3148, pp. 222–229. [Google Scholar]
- Yan, L.; Buenaventura, E.; Pape, T.; Kutty, S.N.; Bayless, K.M.; Zhang, D. A Phylotranscriptomic Framework for Flesh Fly Evolution (Diptera, Calyptratae, Sarcophagidae). Cladistics 2021, 37, 540–558. [Google Scholar] [CrossRef]
- Buenaventura, E.; Szpila, K.; Cassel, B.K.; Wiegmann, B.M.; Pape, T. Anchored Hybrid Enrichment Challenges the Traditional Classification of Flesh Flies (Diptera: Sarcophagidae). Syst. Entomol. 2020, 45, 281–301. [Google Scholar] [CrossRef] [Green Version]
- Pape, T. Catalogue of the Sarcophagidae of the World; Associated Publishers: Gainesville, FL, USA, 1996; Volume 8, pp. 1–558. [Google Scholar]
- Buenaventura, E.; Pape, T. Phylogeny, Evolution and Male Terminalia Functionality of Sarcophaginae (Diptera: Sarcophagidae). Zool. J. Linn. Soc. 2018, 183, 808–906. [Google Scholar] [CrossRef]
- Piwczyński, M.; Pape, T.; Deja-Sikora, E.; Sikora, M.; Akbarzadeh, K.; Szpila, K. Molecular Phylogeny of Miltogramminae (Diptera: Sarcophagidae): Implications for Classification, Systematics and Evolution of Larval Feeding Strategies. Mol. Phylogenet. Evol. 2017, 116, 49–60. [Google Scholar] [CrossRef]
- Piwczyński, M.; Szpila, K.; Grzywacz, A.; Pape, T. A Large-Scale Molecular Phylogeny of Flesh Flies (Diptera: Sarcophagidae). Syst. Entomol. 2014, 39, 783–799. [Google Scholar] [CrossRef]
- Ren, L.; Shang, Y.; Chen, W.; Meng, F.; Cai, J.; Zhu, G.; Chen, L.; Wang, Y.; Deng, J.; Guo, Y. A Brief Review of Forensically Important Flesh Flies (Diptera: Sarcophagidae). Forensic Sci. Res. 2018, 3, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szpila, K.; Voss, J.G.; Pape, T. A New Dipteran Forensic Indicator in Buried Bodies. Med. Vet. Entomol. 2010, 24, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Jordaens, K.; Sonet, G.; Richet, R.; Dupont, E.; Braet, Y.; Desmyter, S. Identification of Forensically Important Sarcophaga Species (Diptera: Sarcophagidae) Using the Mitochondrial COI Gene. Int. J. Legal Med. 2013, 127, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.J.R.; Farkas, R. Traumatic Myiasis of Humans and Animals. In Contributions to a Manual of Palaearctic Diptera; Papp, L., Darvas, B., Eds.; Science Herald: Budapest, Hungary, 2000; Volume 1, pp. 751–768. [Google Scholar]
- Szpila, K.; Hall, M.J.R.; Wardhana, A.H.; Pape, T. Morphology of the First Instar Larva of Obligatory Traumatic Myiasis Agents (Diptera: Calliphoridae, Sarcophagidae). Parasitol. Res. 2014, 113, 1629–1640. [Google Scholar] [CrossRef] [Green Version]
- Sivinski, J.; Marshall, S.; Petersson, E. Kleptoparasitism and Phoresy in the Diptera. Florida Entomol. 1999, 82, 179–197. [Google Scholar] [CrossRef]
- Iyengar, E.V. Kleptoparasitic Interactions throughout the Animal Kingdom and a Re-Evaluation, Based on Participant Mobility, of the Conditions Promoting the Evolution of Kleptoparasitism. Biol. J. Linn. Soc. 2008, 93, 745–762. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.J.R.; Wall, R.L.; Stevens, J.R. Traumatic Myiasis: A Neglected Disease in a Changing World. Annu. Rev. Entomol. 2016, 61, 159–176. [Google Scholar] [CrossRef]
- Kuhlmann, U. Biology and Predation Rate of the Sarcophagid Fly, Agria mamillata a Predator of European Small Ermine Moths. Int. J. Pest Manag. 1995, 41, 67–73. [Google Scholar] [CrossRef]
- Kurahashi, H. The Systematic Position of Nemoraea cicadina Kato, 1943, a Parasite of the Cicada, Tanna japonensis Distant. Jpn. J. Syst. Entomol. 1996, 2, 109–113. [Google Scholar]
- Hall, M.J.R.; Adams, Z.J.O.; Wyatt, N.P.; Testa, J.M.; Edge, W.; Nikolausz, M.; Farkas, R.; Ready, P.D. Morphological and Mitochondrial DNA Characters for Identification and Phylogenetic Analysis of the Myiasis-Causing Flesh Fly Wohlfahrtia magnifica and its Relatives, with a Description of Wohlfahrtia monegrosensis sp. n. Wyatt & Hall. Med. Vet. Entomol. 2009, 23, 59–71. [Google Scholar] [CrossRef]
- Hall, M.; Wall, R. Myiasis of Humans and Domestic Animals. Adv. Parasitol. 1995, 35, 257–334. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect Mitochondrial Genomics: Implications for Evolution and Phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Yan, L.; Zhang, M.; Chu, H.; Cao, J.; Li, K.; Hu, D.; Pape, T. Phylogenetic Inference of Calyptrates, with the First Mitogenomes for Gasterophilinae (Diptera: Oestridae) and Paramacronychiinae (Diptera: Sarcophagidae). Int. J. Biol. Sci. 2016, 12, 489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junqueira, A.C.M.; Azeredo-Espin, A.M.L.; Paulo, D.F.; Marinho, M.A.T.; Tomsho, L.P.; Drautz-Moses, D.I.; Purbojati, R.W.; Ratan, A.; Schuster, S.C. Large-Scale Mitogenomics Enables Insights into Schizophora (Diptera) Radiation and Population Diversity. Sci. Rep. 2016, 6, 21762. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Pape, T.; Elgar, M.A.; Gao, Y.; Zhang, D. Evolutionary History of Stomach Bot Flies in the Light of Mitogenomics. Syst. Entomol. 2019, 44, 797–809. [Google Scholar] [CrossRef]
- Du, Z.; Hasegawa, H.; Cooley, J.R.; Simon, C.; Yoshimura, J.; Cai, W.; Sota, T.; Li, H. Mitochondrial Genomics Reveals Shared Phylogeographic Patterns and Demographic History among Three Periodical Cicada Species Groups. Mol. Biol. Evol. 2019, 36, 1187–1200. [Google Scholar] [CrossRef]
- Du, Z.; Wu, Y.; Chen, Z.; Cao, L.; Ishikawa, T.; Kamitani, S.; Sota, T.; Song, F.; Tian, L.; Cai, W.; et al. Global Phylogeography and Invasion History of the Spotted Lanternfly Revealed by Mitochondrial Phylogenomics. Evol. Appl. 2021, 14, 915–930. [Google Scholar] [CrossRef]
- Li, H.; Leavengood, J.M.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P.; Liu, J.; Zhou, X.; Cai, W. Mitochondrial Phylogenomics of Hemiptera Reveals Adaptive Innovations Driving the Diversification of True Bugs. Proc. R. Soc. B Biol. Sci. 2017, 284, 20171223. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Liu, H.; Ge, X.; Yang, G.; Xie, G.; Yang, Y. A Mitochondrial Genome Phylogeny of Cleridae (Coleoptera, Cleroidea). Insects 2022, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, D. Mitochondrial Genomes Provide New Phylogenetic and Evolutionary Insights into Psilidae (Diptera: Brachycera). Insects 2022, 13, 518. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Li, H.; Liu, G.H.; Wang, W.; James, P.; Colwell, D.D.; Tran, A.; Gong, S.; Cai, W.; Shao, R. Mitochondrial Genome Fragmentation Unites the Parasitic Lice of Eutherian Mammals. Syst. Biol. 2019, 68, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, H.; Song, F.; Zhao, Y.; Wilson, J.J.; Cai, W. Higher-Level Phylogeny and Evolutionary History of Pentatomomorpha (Hemiptera: Heteroptera) Inferred from Mitochondrial Genome Sequences. Syst. Entomol. 2019, 44, 810–819. [Google Scholar] [CrossRef]
- Wei, S.J.; Shi, M.; Chen, X.X.; Sharkey, M.J.; van Achterberg, C.; Ye, G.Y.; He, J.H. New Views on Strand Asymmetry in Insect Mitochondrial Genomes. PLoS ONE 2010, 5, e12708. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Li, H.X.; Yu, X.F.; Yang, M.F. Comparative Analysis of Mitochondrial Genomes among Twelve Sibling Species of the Genus Atkinsoniella Distant, 1908 (Hemiptera: Cicadellidae: Cicadellinae) and Phylogenetic Analysis. Insects 2022, 13, 254. [Google Scholar] [CrossRef] [PubMed]
- Crampton-Platt, A.; Timmermans, M.J.T.N.; Gimmel, M.L.; Kutty, S.N.; Cockerill, T.D.; Vun Khen, C.; Vogler, A.P. Soup to Tree: The Phylogeny of Beetles Inferred by Mitochondrial Metagenomics of a Bornean Rainforest Sample. Mol. Biol. Evol. 2015, 32, 2302–2316. [Google Scholar] [CrossRef]
- Ye, Z.M. Sarcophagidae. In Key to the Common Flies of China; Fan, Z.D., Ed.; Science Press: Beijing, China, 1992; pp. 580–719. [Google Scholar]
- Xue, W.Q.; Chao, C.M. Flies of China; Liaoning Science and Technology Press: Shenyang, China, 1996; Volume 2. [Google Scholar]
- Yan, L.; Xu, W.; Zhang, D.; Li, J. Comparative Analysis of the Mitochondrial Genomes of Flesh Flies and Their Evolutionary Implication. Int. J. Biol. Macromol. 2021, 174, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Read Trimming Tool for Illumina NGS Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de Novo Assembler for Single-Cell and Metagenomic Sequencing Data with Highly Uneven Depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Xia, X.; Xie, Z. DAMBE: Software Package for Data Analysis in Molecular Biology and Evolution. J. Hered. 2001, 92, 371–373. [Google Scholar] [CrossRef] [Green Version]
- Jermiin, L.; Ott, M. SymTest Version 2.0.47. 2017. Available online: https://github.com/ottmi/symt (accessed on 10 June 2022).
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation Software for the Fast Assembly of Multi-Gene Datasets with Character Set and Codon Information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v4: Recent Updates and New Developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.; Zhang, M.; Gao, Y.; Pape, T.; Zhang, D. First Mitogenome for the Subfamily Miltogramminae (Diptera: Sarcophagidae) and Its Phylogenetic Implications. Eur. J. Entomol. 2017, 114, 422–429. [Google Scholar] [CrossRef]
- Ren, L.; Zhang, X.; Li, Y.; Shang, Y.; Chen, S.; Wang, S.; Qu, Y.; Cai, J.; Guo, Y. Comparative Analysis of Mitochondrial Genomes among the Subfamily Sarcophaginae (Diptera: Sarcophagidae) and Phylogenetic Implications. Int. J. Biol. Macromol. 2020, 161, 214–222. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. TRNA Punctuation Model of RNA Processing in Human Mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological Identifications through DNA Barcodes. Proc. R. Soc. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Demari-Silva, B.; Foster, P.G.; de Oliveira, T.M.P.; Bergo, E.S.; Sanabani, S.S.; Pessôa, R.; Sallum, M.A.M. Mitochondrial Genomes and Comparative Analyses of Culex camposi, Culex coronator, Culex usquatus and Culex usquatissimus (Diptera:Culicidae), Members of the Coronator Group. BMC Genom. 2015, 16, 831. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Dai, W.; Dietrich, C.H. Mitochondrial Genomic Variation and Phylogenetic Relationships of Three Groups in the Genus Scaphoideus (Hemiptera: Cicadellidae: Deltocephalinae). Sci. Rep. 2017, 7, 16908. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.K.; Sykes, G.; King, S.; Cottrill, K.; Ivanova, N.V.; Hanner, R.; Ikonomi, P. Species Identification in Cell Culture: A Two-Pronged Molecular Approach. Vitr. Cell. Dev. Biol.-Anim. 2007, 43, 344–351. [Google Scholar] [CrossRef]
- Ma, L.; Liu, F.; Chiba, H.; Yuan, X. The Mitochondrial Genomes of Three Skippers: Insights into the Evolution of the Family Hesperiidae (Lepidoptera). Genomics 2020, 112, 432–441. [Google Scholar] [CrossRef]
- Verves, Y.G. To the Knowledge of the Subfamilies of the Sarcophagidae (Diptera). Int. J. Dipterol. Res. 1998, 9, 243–244. [Google Scholar]
- Zhao, Z.; Su, T.; Chesters, D.; Wang, S.; Ho, S.Y.W.; Zhu, C.; Chen, X.; Zhang, C. The Mitochondrial Genome of Elodia flavipalpis Aldrich (Diptera: Tachinidae) and the Evolutionary Timescale of Tachinid Flies. PLoS ONE 2013, 8, e61814. [Google Scholar] [CrossRef] [PubMed]
- Hassanin, A. Phylogeny of Arthropoda Inferred from Mitochondrial Sequences: Strategies for Limiting the Misleading Effects of Multiple Changes in Pattern and Rates of Substitution. Mol. Phylogenet. Evol. 2006, 38, 100–116. [Google Scholar] [CrossRef] [PubMed]
- Pons, J.; Ribera, I.; Bertranpetit, J.; Balke, M. Nucleotide Substitution Rates for the Full Set of Mitochondrial Protein-Coding Genes in Coleoptera. Mol. Phylogenet. Evol. 2010, 56, 796–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Subfamily | Genus | Subgenus | Species | Accession No. |
|---|---|---|---|---|---|
| Sarcophagidae | Miltogramminae | Mesomelena | mesomelaena | KY003227 | |
| Miltogramma | oestracea | MW556020 | |||
| Phrosinella | nasuta | MW546340 | |||
| Protomiltogramma | cincta | NC_063942 | |||
| Sarcotachina | subcylindrica | MW556014 | |||
| Senotainia | albifrons | MW556016 | |||
| Sphecapatoclea | sp. | MW556015 | |||
| Taxigramma | karakulensis * | ON375459 | |||
| Paramacronychiinae | Agria | mihalyii * | ON360967 | ||
| Goniophyto | honshuensis | MW556017 | |||
| Sarcophila | latifrons | MW556018 | |||
| Wohlfahrtia | balassogloi * | ON411642 | |||
| Wohlfahrtia | cheni | MW556019 | |||
| Wohlfahrtia | fedtschenkoi * | ON411643 | |||
| Wohlfahrtia | magnifica | KU578263 | |||
| Sarcophaginae | Blaesoxipha | lapidosa * | OM640654 | ||
| Boettcheria | bisetosa | KT272844 | |||
| latisterna | KT272848 | ||||
| Sarcophaga | Bellieriomima | diminuta | MT017719 | ||
| Bellieriomima | graciliforceps | MT017712 | |||
| Bellieriomima | josephi | MT017711 | |||
| Bellieriomima | pterygota | MK820722 | |||
| Bercaea | africa | KM881633 | |||
| Boettcherisca | formosensis | MF688648 | |||
| Boettcherisca | peregrina | KF921296 | |||
| Helicophagella | melanura | KP091687 | |||
| Heteronychia | depressifrons | MT017709 | |||
| Heteronychia | pauciseta | NC_053729 | |||
| Heteronychia | plotnikovi | MT017720 | |||
| Heteronychia | shnitnikovi | MT017730 | |||
| Heteronychia | tsinanensis | NC_057591 | |||
| Kozlovea | cetu | MT017725 | |||
| Kramerea | schuetzei | MT017726 | |||
| Liopygia | crassipalpis | KC005711 | |||
| Liopygia | ruficornis | MH937749 | |||
| Liosarcophaga | aegyptica | MT017732 | |||
| Liosarcophaga | brevicornis | NC_047404 | |||
| Liosarcophaga | dux | MH879759 | |||
| Liosarcophaga | jacobsoni | MT017723 | |||
| Liosarcophaga | kanoi | MT476487 | |||
| Liosarcophaga | portschinskyi | KM287570 | |||
| Liosarcophaga | tuberosa | MK820723 | |||
| Neobellieria | bullata | KT272859 | |||
| Pandelleisca | kawayuensis | MT017713 | |||
| Pandelleisca | pingi | MT017728 | |||
| Pandelleisca | scopariiformis | MT476486 | |||
| Pandelleisca | similis | KM287431 | |||
| Parasarcophaga | albiceps | KT444443 | |||
| Parasarcophaga | macroauriculata | MT017718 | |||
| Parasarcophaga | misera | MF133500 | |||
| Phallocheira | minor | MT017727 | |||
| Pseudothyrsocnema | caudagalli | MK820721 | |||
| Robineauella | anchoriformis | MT017716 | |||
| Sarcophaga | carnaria | MT017710 | |||
| Sarcorohdendorfia | antilope | MH540748 | |||
| Sarcorohdendorfia | gracilior | MW531675 | |||
| Sarcorohdendorfia | impatiens | JN859549 | |||
| Seniorwhitea | princeps | MH981944 | |||
| Sinonipponia | hervebazini | MT017708 | |||
| Thyrsocnema | kentejana | MT017714 | |||
| Ravinia | pernix | KM676414 | |||
| Oxysarcodexia | avuncula | MH879754 | |||
| Oxysarcodexia | terminalis | MH879757 | |||
| Oxysarcodexia | thornax | MH879765 | |||
| Oxysarcodexia | varia | MH879764 | |||
| Peckia | australis | MH879762 | |||
| Peckia | collusor | MH879763 | |||
| Peckia | resona | MH879761 | |||
| Calliphoridae | Calliphora | vomitoria | KT444440 | ||
| Anthomyiidae | Delia | antiqua | NC_028226 | ||
| Fanniidae | Fannia | scalaris | MT017706 | ||
| Muscidae | Musca | domestica | KM200723 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, J.; Xu, W.; Huang, X.; Zhang, D.; Yan, L.; Pape, T. Comparative Mitogenomics of Flesh Flies: Implications for Phylogeny. Insects 2022, 13, 718. https://doi.org/10.3390/insects13080718
Shang J, Xu W, Huang X, Zhang D, Yan L, Pape T. Comparative Mitogenomics of Flesh Flies: Implications for Phylogeny. Insects. 2022; 13(8):718. https://doi.org/10.3390/insects13080718
Chicago/Turabian StyleShang, Jin, Wentian Xu, Xiaofang Huang, Dong Zhang, Liping Yan, and Thomas Pape. 2022. "Comparative Mitogenomics of Flesh Flies: Implications for Phylogeny" Insects 13, no. 8: 718. https://doi.org/10.3390/insects13080718