
Exploring Large-Scale Patterns of Genetic Variation in the COI Gene among Insecta: Implications for DNA Barcoding and Threshold-Based Species Delimitation Studies

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Filtering
2.2. Analysis at the Species Level
2.3. Analysis at the Genus Level
3. Results
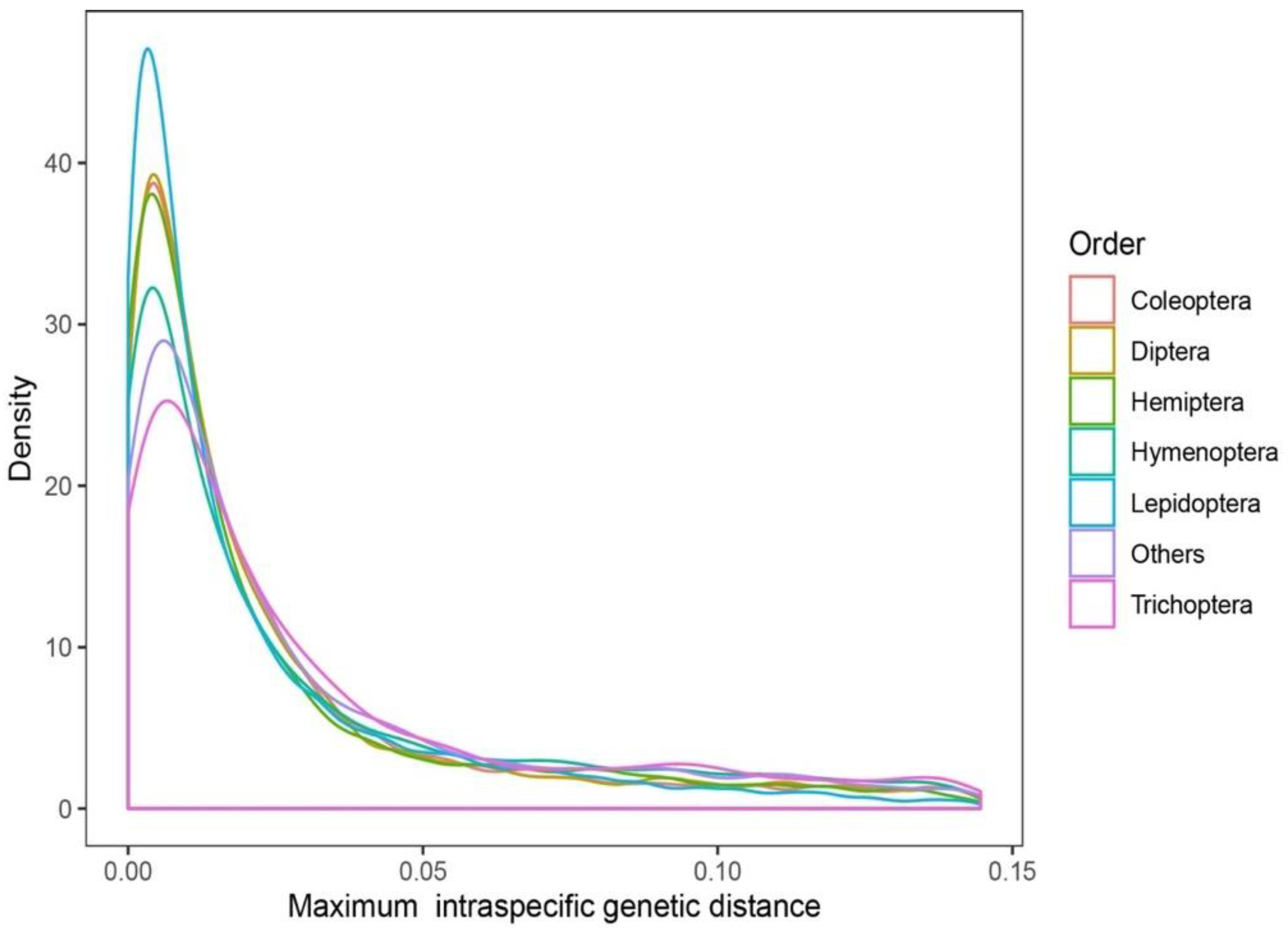
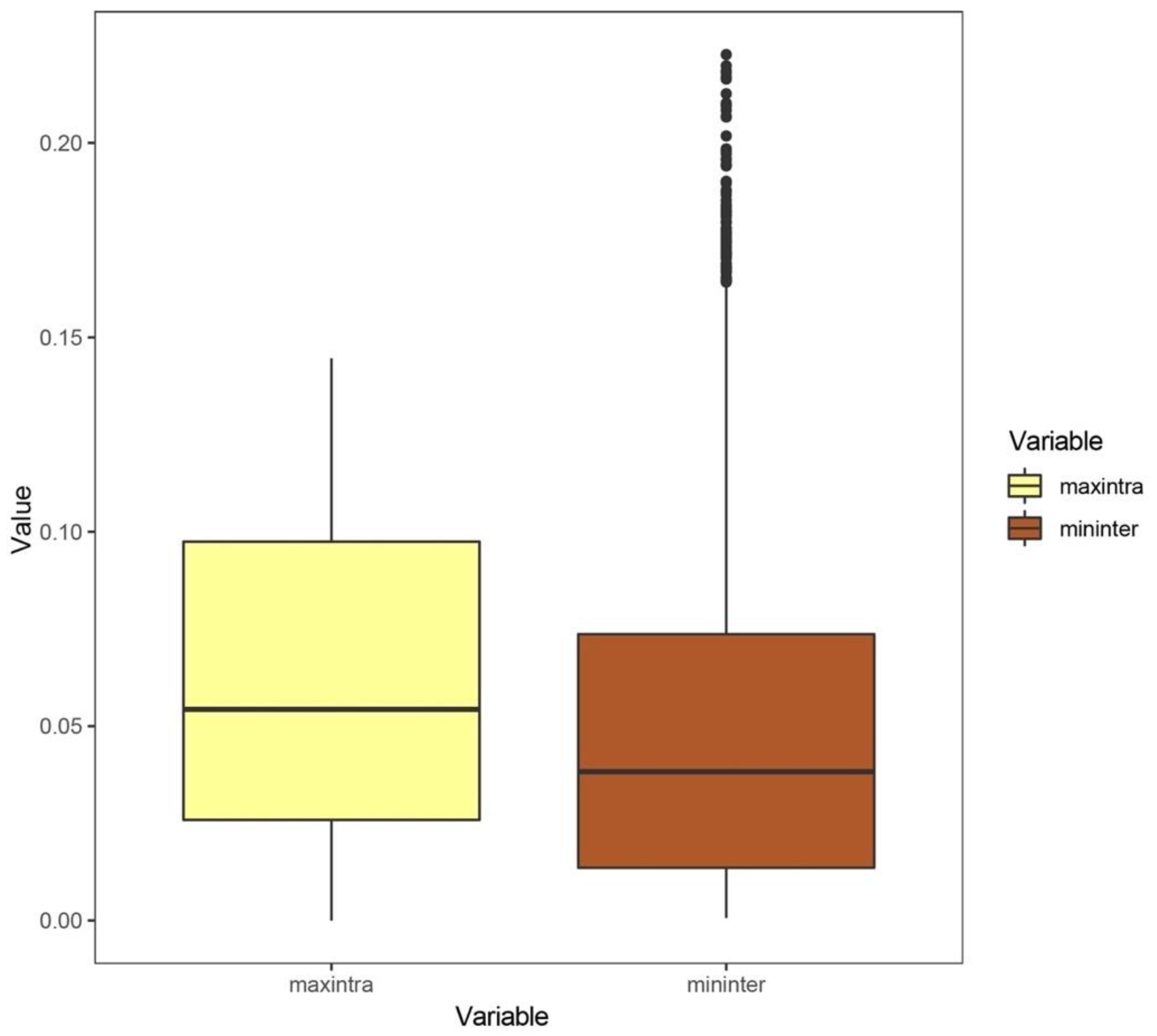
3.1. Analysis Results at the Species Level
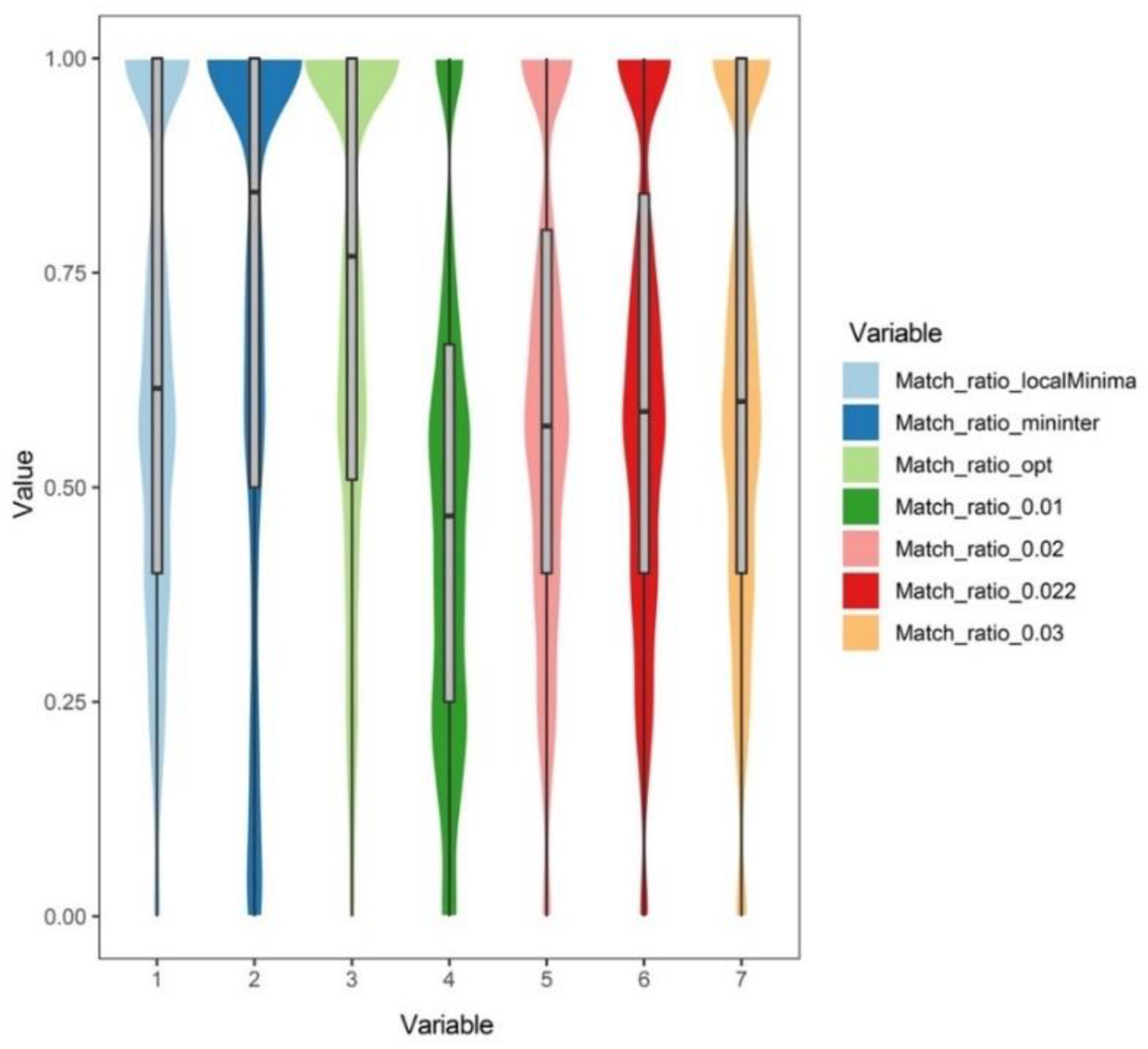
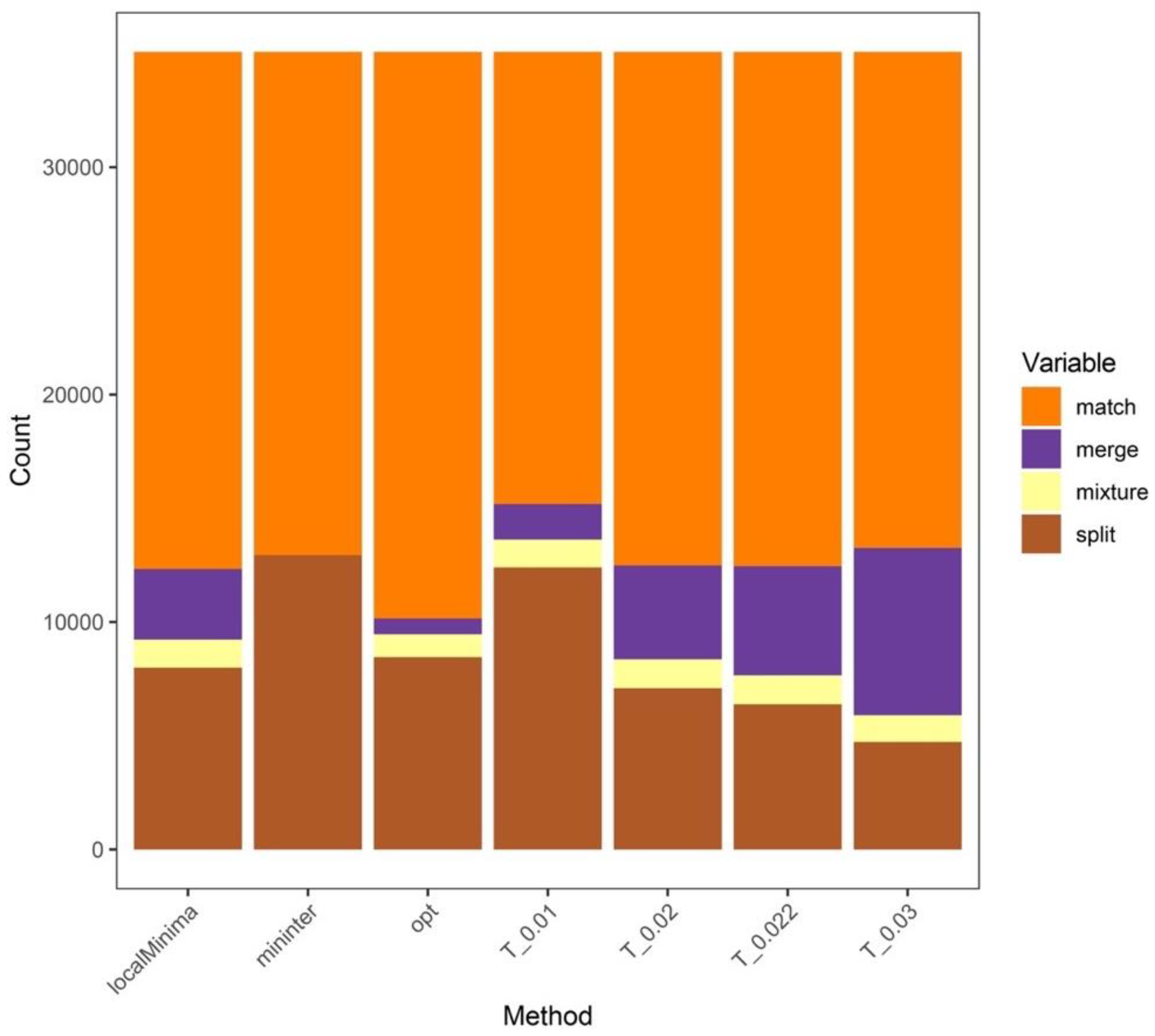
3.2. Analysis Results at the Genus Level
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Z.Q. Animal biodiversity: An introduction to higher-level classification and taxonomic richness. Zootaxa 2011, 3148, 7–12. [Google Scholar] [CrossRef]
- Stork, N.E. How Many Species of Insects and Other Terrestrial Arthropods Are There on Earth? Annu. Rev. Entomol. 2018, 63, 31–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stork, N.E.; McBroom, J.; Gely, C.; Hamilton, A.J. New approaches narrow global species estimates for beetles, insects, and terrestrial arthropods. Proc. Natl. Acad. Sci. USA 2015, 112, 7519–7523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foottit, R.G.; Adler, P.H. Insect Biodiversity: Science and Society; Wiley-Blackwell: Chichester, UK, 2009. [Google Scholar]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G.; et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Barraclough, T.G. Delimiting species using single-locus data and the Generalized Mixed Yule Coalescent approach: A revised method and evaluation on simulated data sets. Syst. Biol. 2013, 62, 707–724. [Google Scholar] [CrossRef] [Green Version]
- Ratnasingham, S.; Hebert, P.D.N. A DNA-based registry for all animal species: The Barcode Index Number (BIN) system. PLoS ONE 2013, 8, e66213. [Google Scholar] [CrossRef] [Green Version]
- Machida, R.J.; Leray, M.; Ho, S.-L.; Knowlton, N. Metazoan mitochondrial gene sequence reference datasets for taxonomic assignment of environmental samples. Sci. Data 2017, 4, 170027. [Google Scholar] [CrossRef]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; deWaard, J.R. Biological identifications through DNA barcodes. Proc. Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Ashfaq, M.; Sabir, J.S.M.; El-Ansary, H.O.; Perez, K.; Levesque-Beaudin, V.; Khan, A.M.; Rasool, A.; Gallant, C.; Addesi, J.; Hebert, P.D.N. Insect diversity in the Saharo-Arabian region: Revealing a little-studied fauna by DNA barcoding. PLoS ONE 2018, 13, e0199965. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, A.; Assandri, G.; Maggioni, D.; Ramazzotti, F.; Baroni, D.; Bazzi, G.; Chiandetti, I.; Corso, A.; Ferri, V.; Galuppi, M.; et al. Italian odonates in the Pandora’s box: A comprehensive DNA barcoding inventory shows taxonomic warnings at the Holarctic scale. Mol. Ecol. Resour. 2021, 21, 183–200. [Google Scholar] [CrossRef]
- Hajibabaei, M.; Janzen, D.H.; Burns, J.M.; Hallwachs, W.; Hebert, P.D.N. DNA barcodes distinguish species of tropical Lepidoptera. Proc. Natl. Acad. Sci. USA 2006, 103, 968–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawlitschek, O.; Moriniere, J.; Lehmann, G.U.C.; Lehmann, A.W.; Kropf, M.; Dunz, A.; Glaw, F.; Detcharoen, M.; Schmidt, S.; Hausmann, A.; et al. DNA barcoding of crickets, katydids and grasshoppers (Orthoptera) from Central Europe with focus on Austria, Germany and Switzerland. Mol. Ecol. Resour. 2017, 17, 1037–1053. [Google Scholar] [CrossRef] [PubMed]
- Hendrich, L.; Moriniere, J.; Haszprunar, G.; Hebert, P.D.; Hausmann, A.; Kohler, F.; Balke, M. A comprehensive DNA barcode database for Central European beetles with a focus on Germany: Adding more than 3500 identified species to BOLD. Mol. Ecol. Resour. 2015, 15, 795–818. [Google Scholar] [CrossRef] [PubMed]
- Janzen, D.H.; Hajibabaei, M.; Burns, J.M.; Hallwachs, W.; Remigio, E.; Hebert, P.D.N. Wedding biodiversity inventory of a large and complex Lepidoptera fauna with DNA barcoding. Philos. Trans. R. Soc. B 2005, 360, 1835–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janzen, D.H.; Hallwachs, W.; Blandin, P.; Burns, J.M.; Cadiou, J.M.; Chacon, I.; Dapkey, T.; Deans, A.R.; Epstein, M.E.; Espinoza, B.; et al. Integration of DNA barcoding into an ongoing inventory of complex tropical biodiversity. Mol. Ecol. Resour. 2009, 9 (Suppl. 1), 1–26. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Duwal, R.K.; Lee, S. COI barcoding of true bugs (Insecta, Heteroptera). Mol. Ecol. Resour. 2011, 11, 266–270. [Google Scholar] [CrossRef]
- Meier, R.; Shiyang, K.; Vaidya, G.; Ng, P.K.L. DNA barcoding and taxonomy in diptera: A tale of high intraspecific variability and low identification success. Syst. Biol. 2006, 55, 715–728. [Google Scholar] [CrossRef] [Green Version]
- Moriniere, J.; Balke, M.; Doczkal, D.; Geiger, M.F.; Hardulak, L.A.; Haszprunar, G.; Hausmann, A.; Hendrich, L.; Regalado, L.; Rulik, B.; et al. A DNA barcode library for 5200 German flies and midges (Insecta: Diptera) and its implications for metabarcoding-based biomonitoring. Mol. Ecol. Resour. 2019, 19, 900–928. [Google Scholar] [CrossRef] [Green Version]
- Pentinsaari, M.; Hebert, P.D.; Mutanen, M. Barcoding beetles: A regional survey of 1872 species reveals high identification success and unusually deep interspecific divergences. PLoS ONE 2014, 9, e108651. [Google Scholar] [CrossRef] [Green Version]
- Schmid-Egger, C.; Straka, J.; Ljubomirov, T.; Blagoev, G.A.; Moriniere, J.; Schmidt, S. DNA barcodes identify 99 per cent of apoid wasp species (Hymenoptera: Ampulicidae, Crabronidae, Sphecidae) from the Western Palearctic. Mol. Ecol. Resour. 2019, 19, 476–484. [Google Scholar] [CrossRef]
- Schmidt, S.; Taeger, A.; Moriniere, J.; Liston, A.; Blank, S.M.; Kramp, K.; Kraus, M.; Schmidt, O.; Heibo, E.; Prous, M.; et al. Identification of sawflies and horntails (Hymenoptera, ‘Symphyta’) through DNA barcodes: Successes and caveats. Mol. Ecol. Resour. 2017, 17, 670–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahlhut, J.K.; Fernandez-Triana, J.; Adamowicz, S.J.; Buck, M.; Goulet, H.; Hebert, P.D.N.; Huber, J.T.; Merilo, M.T.; Sheffield, C.S.; Woodcock, T.; et al. DNA barcoding reveals diversity of Hymenoptera and the dominance of parasitoids in a sub-arctic environment. BMC Ecol. 2013, 13, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergsten, J.; Bilton, D.T.; Fujisawa, T.; Elliott, M.; Monaghan, M.T.; Balke, M.; Hendrich, L.; Geijer, J.; Herrmann, J.; Foster, G.N.; et al. The effect of geographical scale of sampling on DNA barcoding. Syst. Biol. 2012, 61, 851–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.G.; Lv, M.H.; Yi, W.B.; Zhu, W.B.; Bu, W.J. Species diversity can be overestimated by a fixed empirical threshold: Insights from DNA barcoding of the genus Cletus (Hemiptera: Coreidae) and the meta-analysis of COI data from previous phylogeographical studies. Mol. Ecol. Resour. 2017, 17, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.N.; Ratnasingham, S.; deWaard, J.R. Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proc. Biol. Sci. 2003, 270 (Suppl. 1), S96–S99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratnasingham, S.; Hebert, P.D.N. BOLD: The Barcode of Life Data system (www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.A.; Fernandez-Triana, J.L.; Eveleigh, E.; Gomez, J.; Guclu, C.; Hallwachs, W.; Hebert, P.D.N.; Hrcek, J.; Huber, J.T.; Janzen, D.; et al. DNA barcoding and the taxonomy of Microgastrinae wasps (Hymenoptera, Braconidae): Impacts after 8years and nearly 20,000 sequences. Mol. Ecol. Resour. 2013, 13, 168–176. [Google Scholar] [CrossRef]
- Burns, J.M.; Janzen, D.H.; Hajibabaei, M.; Hallwachs, W.; Hebert, P.D. DNA barcodes and cryptic species of skipper butterflies in the genus Perichares in Area de Conservacion Guanacaste, Costa Rica. Proc. Natl. Acad. Sci. USA 2008, 105, 6350–6355. [Google Scholar] [CrossRef] [Green Version]
- Hebert, P.D.N.; Penton, E.H.; Burns, J.M.; Janzen, D.H.; Hallwachs, W. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. USA 2004, 101, 14812–14817. [Google Scholar] [CrossRef] [Green Version]
- Hou, G.; Chen, W.T.; Lu, H.S.; Cheng, F.; Xie, S.G. Developing a DNA barcode library for perciform fishes in the South China Sea: Species identification, accuracy and cryptic diversity. Mol. Ecol. Resour. 2018, 18, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Hsu, T.H.; Ning, Y.; Gwo, J.C.; Zeng, Z.N. DNA barcoding reveals cryptic diversity in the peanut worm Sipunculus nudus. Mol. Ecol. Resour. 2013, 13, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Lyra, M.L.; Haddad, C.F.B.; de Azeredo-Espin, A.M.L. Meeting the challenge of DNA barcoding Neotropical amphibians: Polymerase chain reaction optimization and new COI primers. Mol. Ecol. Resour. 2017, 17, 966–980. [Google Scholar] [CrossRef] [PubMed]
- Muster, C.; Michalik, P. Cryptic diversity in ant-mimic Micaria spiders (Araneae, Gnaphosidae) and a tribute to early naturalists. Zool. Scr. 2020, 49, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Fisher, B.L.; Hebert, P.D. DNA barcoding for effective biodiversity assessment of a hyperdiverse arthropod group: The ants of Madagascar. Philos. Trans. R. Soc. B. 2005, 360, 1825–1834. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Rodriguez, J.J.; Whitfield, J.B.; Deans, A.R.; Janzen, D.H.; Hallwachs, W.; Hebert, P.D.N. Extreme diversity of tropical parasitoid wasps exposed by iterative integration of natural history, DNA barcoding, morphology, and collections. Proc. Natl. Acad. Sci. USA 2008, 105, 12359–12364. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Woodley, N.E.; Janzen, D.H.; Hallwachs, W.; Hebert, P.D. DNA barcodes reveal cryptic host-specificity within the presumed polyphagous members of a genus of parasitoid flies (Diptera: Tachinidae). Proc. Natl. Acad. Sci. USA 2006, 103, 3657–3662. [Google Scholar] [CrossRef] [Green Version]
- Vega-Sánchez, Y.M.; Mendoza-Cuenca, L.F.; González-Rodríguez, A. Complex evolutionary history of the American Rubyspot damselfly, Hetaerina americana (Odonata): Evidence of cryptic speciation. Mol. Phylogenet. Evol. 2019, 139, 106536. [Google Scholar] [CrossRef]
- Vieites, D.R.; Wollenberg, K.C.; Andreone, F.; Kohler, J.; Glaw, F.; Vences, M. Vast underestimation of Madagascar’s biodiversity evidenced by an integrative amphibian inventory. Proc. Natl. Acad. Sci. USA 2009, 106, 8267–8272. [Google Scholar] [CrossRef] [Green Version]
- von Beeren, C.; Maruyama, M.; Kronauer, D.J.C. Cryptic diversity, high host specificity and reproductive synchronization in army ant-associated Vatesus beetles. Mol. Ecol. 2016, 25, 990–1005. [Google Scholar] [CrossRef]
- Meyer, C.P.; Paulay, G. DNA barcoding: Error rates based on comprehensive sampling. PLoS Biol. 2005, 3, 2229–2238. [Google Scholar] [CrossRef] [Green Version]
- Collins, R.A.; Cruickshank, R.H. Known Knowns, Known Unknowns, Unknown Unknowns and Unknown Knowns in DNA Barcoding: A Comment on Dowton et al. Syst. Biol. 2014, 63, 1005–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, T.M.; Hajibabaei, M. Over 2.5 million COI sequences in GenBank and growing. PLoS ONE 2018, 13, e0200177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virgilio, M.; Backeljau, T.; Nevado, B.; De Meyer, M. Comparative performances of DNA barcoding across insect orders. BMC Bioinform. 2010, 11, 206. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, F.M.; Gonçalves, L.T. Borrowing the Pentatomomorpha tome from the DNA barcode library: Scanning the overall performance of cox1 as a tool. J. Zool. Syst. Evol. Res. 2021, 59, 992–1012. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.D.J.; Collins, R.A.; Boyer, S.; Lefort, M.C.; Malumbres-Olarte, J.; Vink, C.J.; Cruickshank, R.H. Spider: An R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Mol. Ecol. Resour. 2012, 12, 562–565. [Google Scholar] [CrossRef]
- Ahrens, D.; Fujisawa, T.; Krammer, H.J.; Eberle, J.; Fabrizi, S.; Vogler, A.P. Rarity and Incomplete Sampling in DNA-Based Species Delimitation. Syst. Biol. 2016, 65, 478–494. [Google Scholar] [CrossRef] [Green Version]
- Blair, C.; Bryson, R.W., Jr. Cryptic diversity and discordance in single-locus species delimitation methods within horned lizards (Phrynosomatidae: Phrynosoma). Mol. Ecol. Resour. 2017, 17, 1168–1182. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Meier, R.; Zhang, G.; Ali, F. The use of mean instead of smallest interspecific distances exaggerates the size of the “barcoding gap” and leads to misidentification. Syst. Biol. 2008, 57, 809–813. [Google Scholar] [CrossRef]
- Srivathsan, A.; Meier, R. On the inappropriate use of Kimura-2-parameter (K2P) divergences in the DNA barcoding literature. Cladistics 2012, 28, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.A.; Boykin, L.M.; Cruickshank, R.H.; Armstrong, K.F. Barcoding’s next top model: An evaluation of nucleotide substitution models for specimen identification. Methods Ecol. Evol. 2012, 3, 457–465. [Google Scholar] [CrossRef]
- Carstens, B.C.; Pelletier, T.A.; Reid, N.M.; Satler, J.D. How to fail at species delimitation. Mol. Ecol. 2013, 22, 4369–4383. [Google Scholar] [CrossRef]
- Collins, R.A.; Cruickshank, R.H. The seven deadly sins of DNA barcoding. Mol. Ecol. Resour. 2013, 13, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Mutanen, M.; Kivela, S.M.; Vos, R.A.; Doorenweerd, C.; Ratnasingham, S.; Hausmann, A.; Huemer, P.; Dinca, V.; van Nieukerken, E.J.; Lopez-Vaamonde, C.; et al. Species-Level Para- and Polyphyly in DNA Barcode Gene Trees: Strong Operational Bias in European Lepidoptera. Syst. Biol. 2016, 65, 1024–1040. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mininter | SN | M_0.01 | M_0.02 | M_0.022 | M_0.03 | M_mininter |
|---|---|---|---|---|---|---|
| ≥0 | 35,068 | 57,950 (65%) | 44,367 (27%) | 42,603 (21%) | 37,698 (7%) | 78,353 (123%) |
| ≥0.01 | 20,714 | 35,243 (70%) | 27,473 (33%) | 26,541 (28%) | 23,976 (16%) | 24,967 (21%) |
| ≥0.02 | 16,037 | 27,653 (72%) | 21,988 (37%) | 21,298 (33%) | 19,476 (21%) | 18,171 (13%) |
| ≥0.03 | 13,034 | 22,909 (76%) | 18,139 (39%) | 17,651 (35%) | 16,474 (26%) | 14,484 (11%) |
| ≥0.04 | 10,454 | 18,584 (78%) | 14,646 (40%) | 14,237 (36%) | 13,278 (27%) | 11,366 (9%) |
| ≥0.05 | 8395 | 15,029 (79%) | 11,803 (41%) | 11,466 (37%) | 10,708 (28%) | 8997 (7%) |
| ≥0.06 | 6706 | 12,196 (82%) | 9521 (42%) | 9256 (38%) | 8627 (29%) | 7093 (6%) |
| ≥0.07 | 5317 | 9810 (85%) | 7633 (44%) | 7406 (39%) | 6908 (30%) | 5569 (5%) |
| ≥0.08 | 4177 | 7706 (84%) | 6015 (44%) | 5842 (40%) | 5457 (31%) | 4326 (4%) |
| ≥0.09 | 3266 | 6093 (87%) | 4724 (45%) | 4589 (41%) | 4273 (31%) | 3365 (3%) |
| ≥0.1 | 2395 | 4456 (86%) | 3456 (44%) | 3356 (40%) | 3131 (31%) | 2447 (2%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Bu, W. Exploring Large-Scale Patterns of Genetic Variation in the COI Gene among Insecta: Implications for DNA Barcoding and Threshold-Based Species Delimitation Studies. Insects 2022, 13, 425. https://doi.org/10.3390/insects13050425
Zhang H, Bu W. Exploring Large-Scale Patterns of Genetic Variation in the COI Gene among Insecta: Implications for DNA Barcoding and Threshold-Based Species Delimitation Studies. Insects. 2022; 13(5):425. https://doi.org/10.3390/insects13050425
Chicago/Turabian StyleZhang, Haiguang, and Wenjun Bu. 2022. "Exploring Large-Scale Patterns of Genetic Variation in the COI Gene among Insecta: Implications for DNA Barcoding and Threshold-Based Species Delimitation Studies" Insects 13, no. 5: 425. https://doi.org/10.3390/insects13050425