
Integrative Proteomic and Phosphoproteomic Analyses Revealed Complex Mechanisms Underlying Reproductive Diapause in Bombus terrestris Queens
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
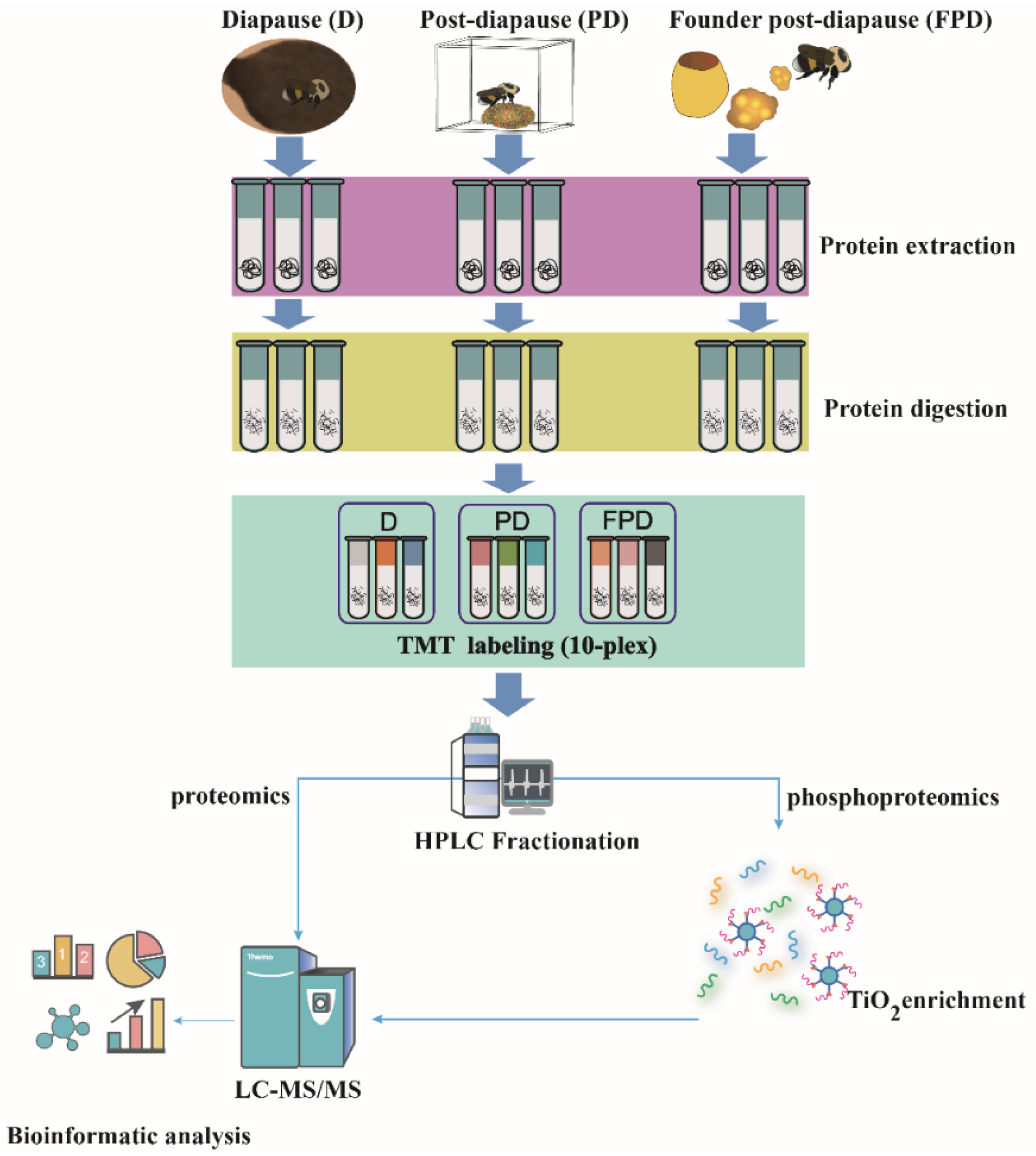
2.1. Bumblebee Sampling
2.2. Protein Extraction and Digestion
2.3. TMT Labeling
2.4. High-pH Peptide Fractionation and Enrichment of Phosphopeptides
2.5. LC-MS/MS Analysis
2.6. Database Search
2.7. Gene Ontology (GO), Protein Domain, Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Annotation, Subcellular Localization, and Mfuzz Analyses
2.8. Motif Analysis
2.9. Kinase Analysis
2.10. PPI Networks
3. Results
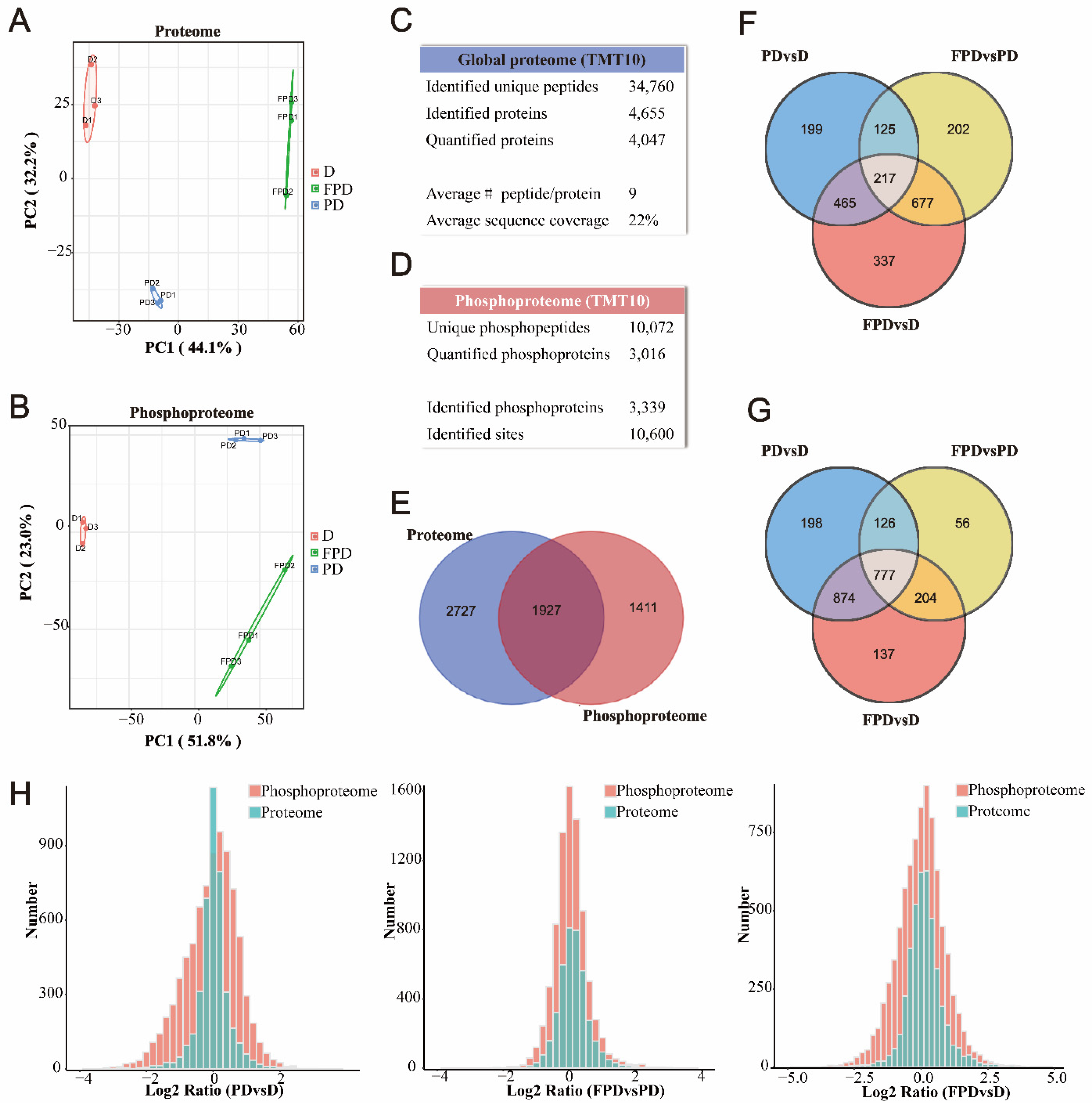
3.1. Quantitative Proteomic and Phosphoproteomic Analyses of B. terrestris Queens in the D, PD, FPD Stages
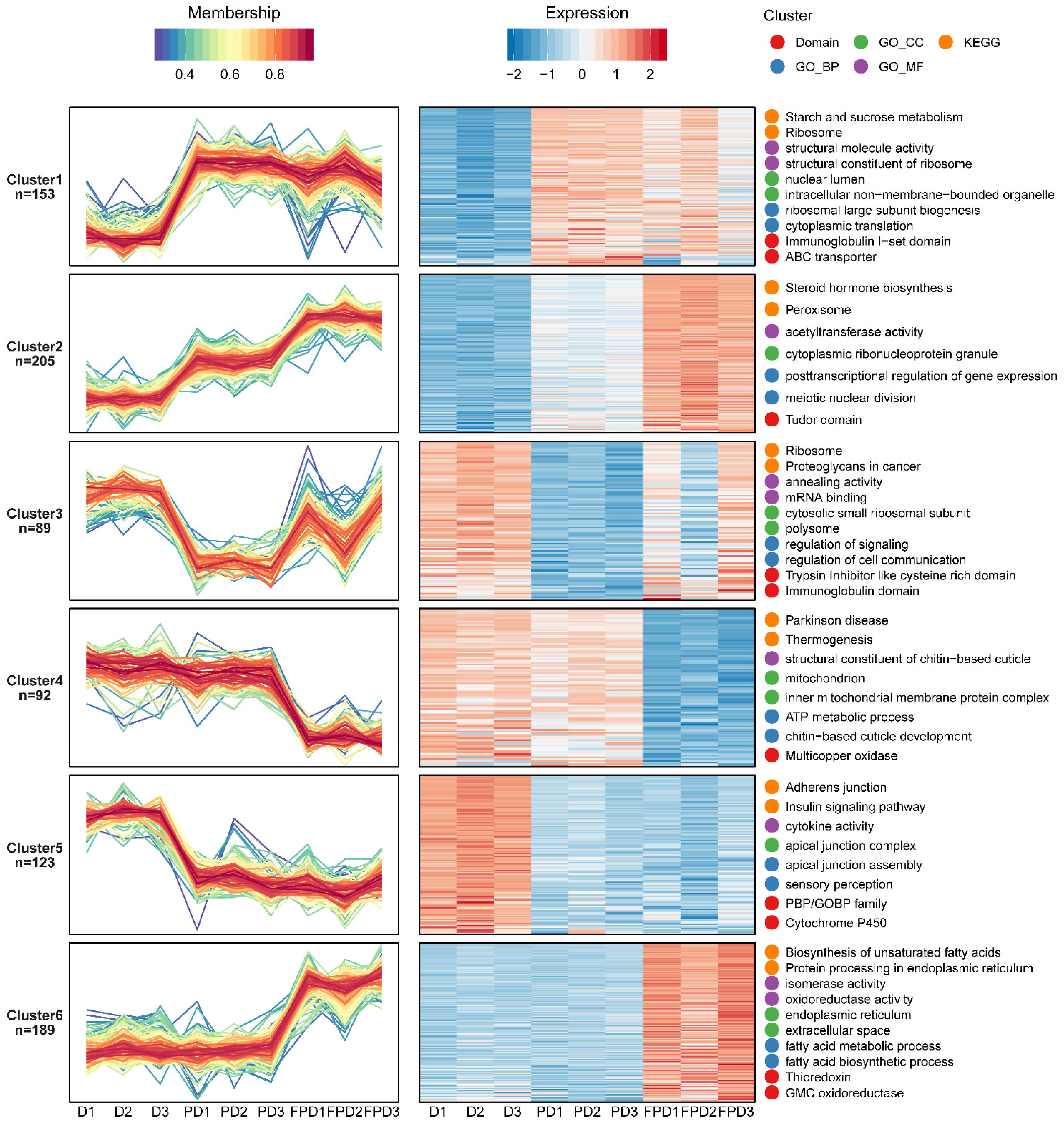
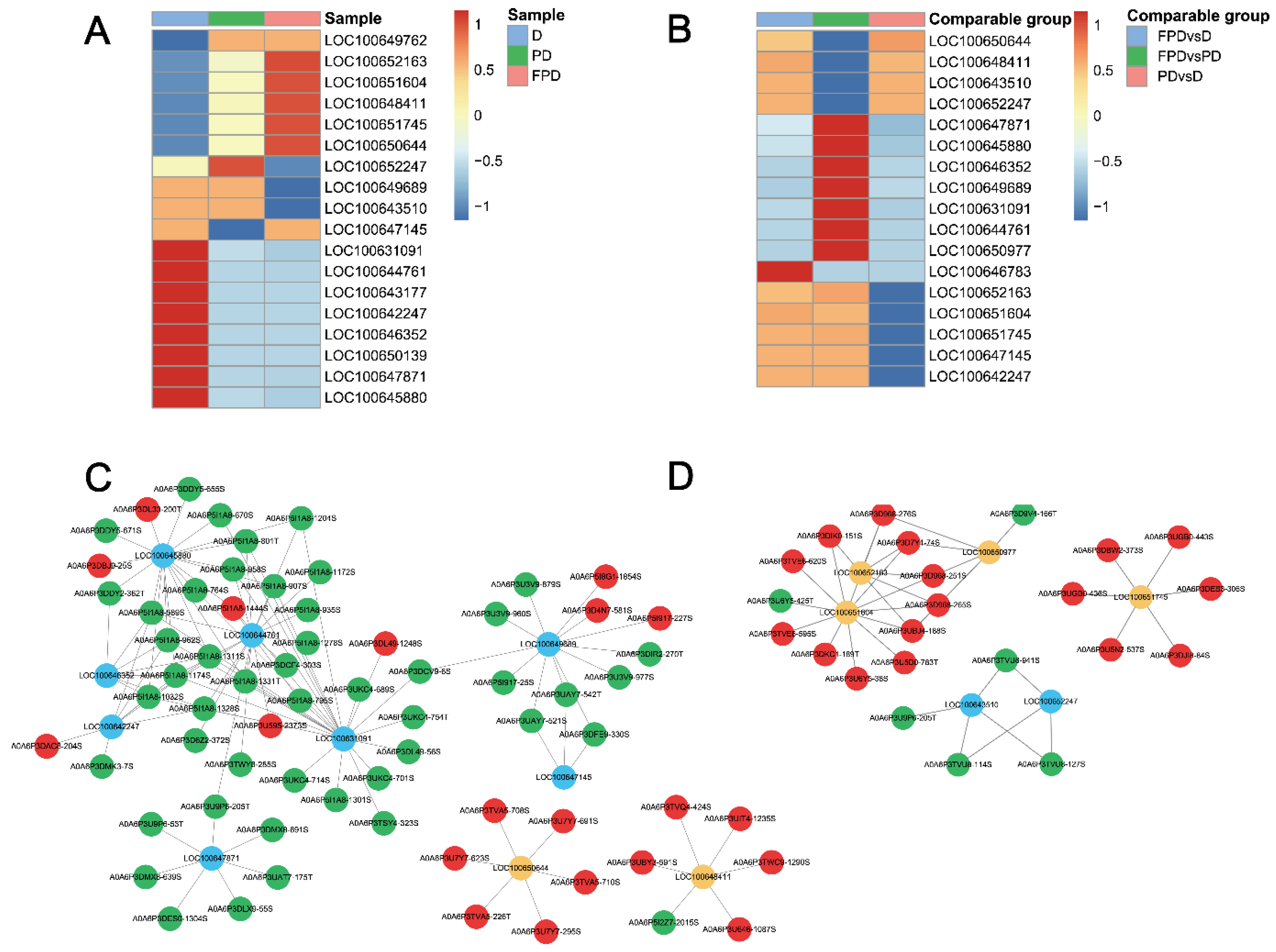
3.2. Expression Profile of Proteins during Different Diapause Stages
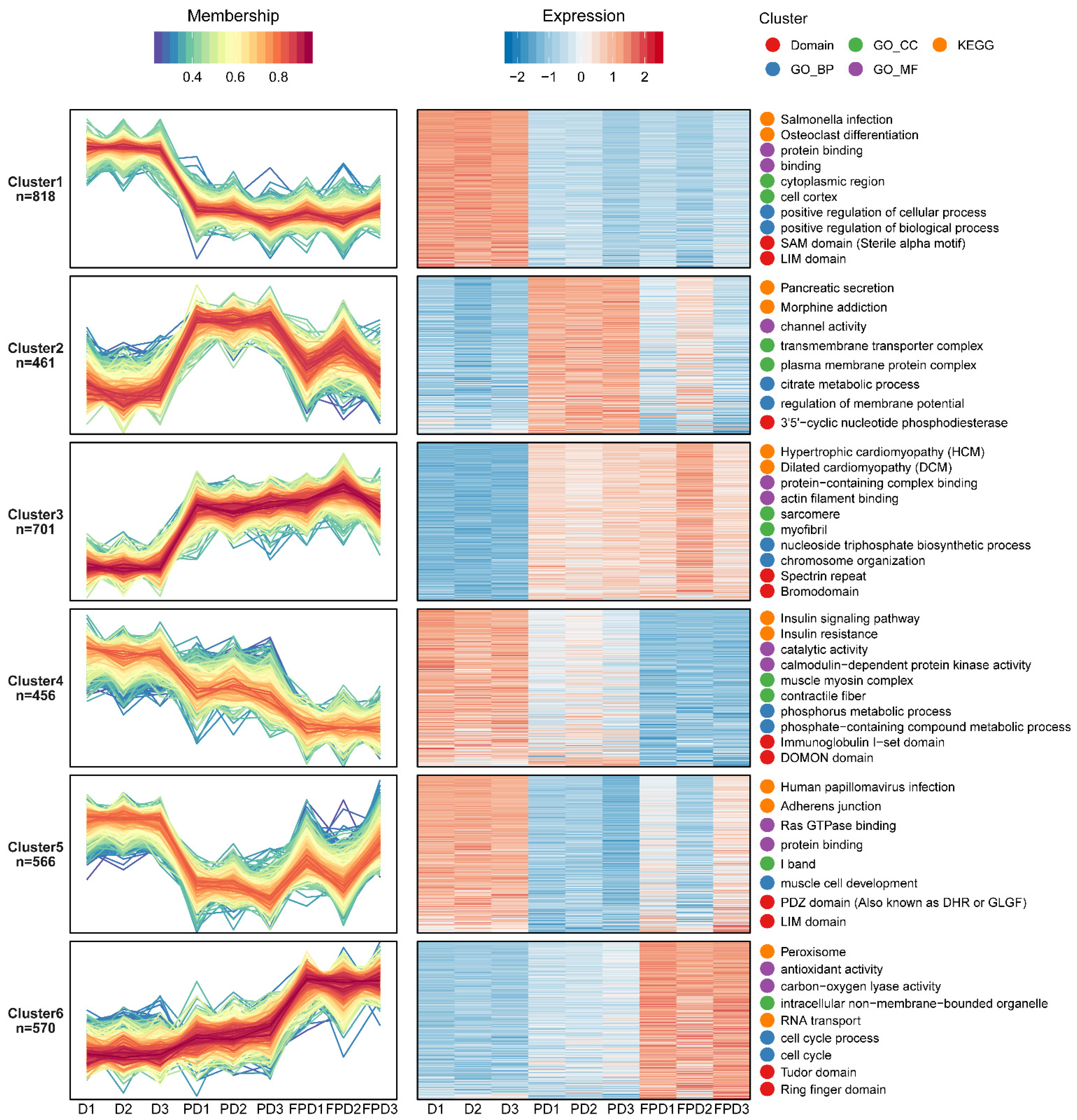
3.3. Functional Analysis of the Identified Phosphoproteins
3.4. Kinase Analysis
3.5. Motif Analysis
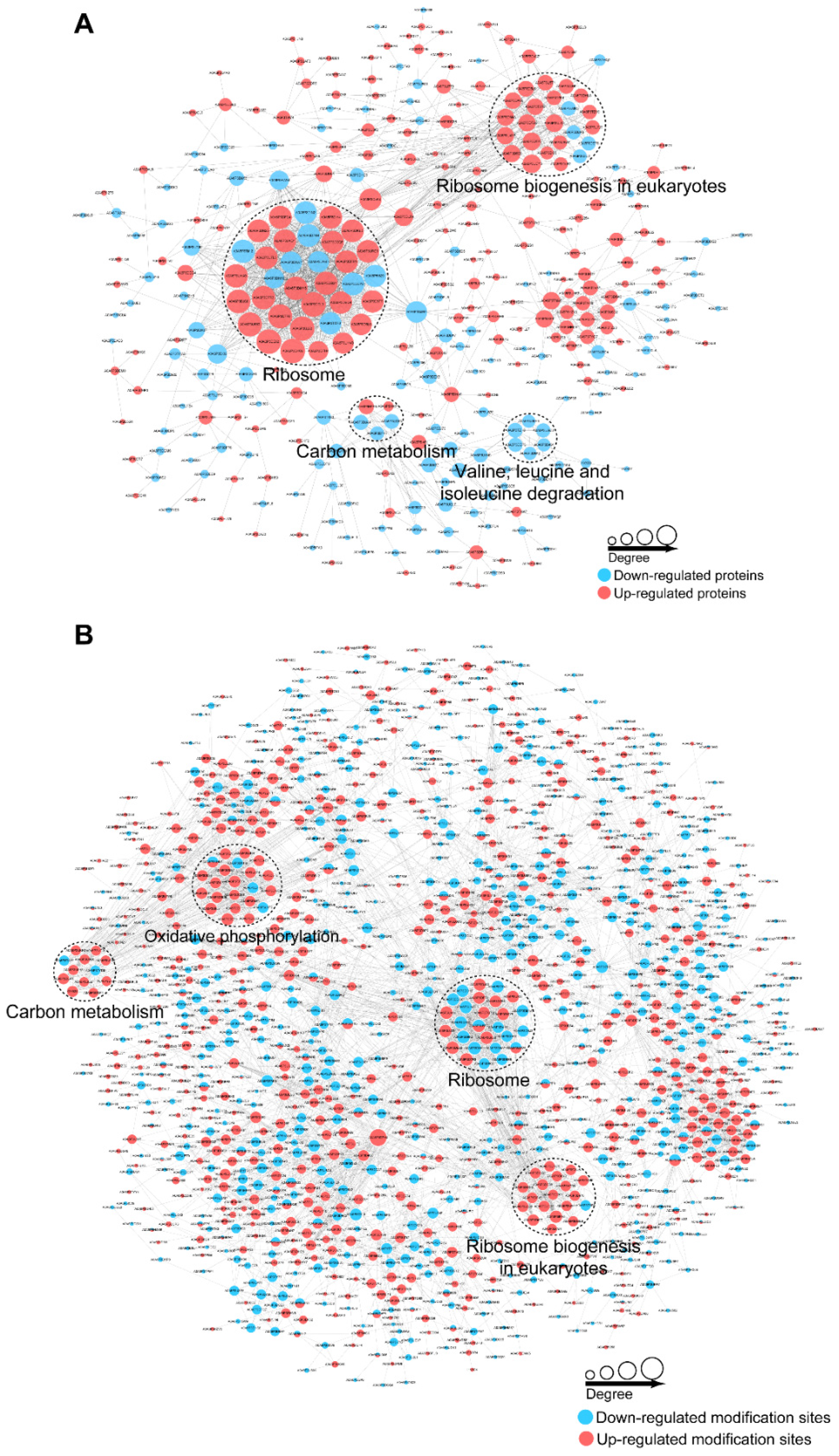
3.6. PPI and Module Analysis of the DEPs and DEPPs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Kostal, V. Eco-physiological phases of insect diapause. J. Insect Physiol. 2006, 52, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Batz, Z.A.; Brent, C.S.; Marias, M.R.; Sugijanto, J.; Armbruster, P.A. Juvenile Hormone III but Not 20-Hydroxyecdysone Regulates the Embryonic Diapause of Aedes albopictus. Front. Physiol. 2019, 10, 1352. [Google Scholar] [CrossRef] [PubMed]
- Storey, K.B.; Storey, J.M. Metabolic rate depression in animals: Transcriptional and translational controls. Biol. Rev. Camb. Philos. Soc. 2004, 79, 207–233. [Google Scholar] [CrossRef] [PubMed]
- Kojic, D.; Popovic, Z.D.; Orcic, D.; Purac, J.; Orcic, S.; Vukasinovic, E.L.; Nikolic, T.V.; Blagojevic, D.P. The influence of low temperature and diapause phase on sugar and polyol content in the European corn borer Ostrinia nubilalis (Hbn.). J. Insect Physiol. 2018, 109, 107–113. [Google Scholar] [CrossRef]
- Moraiti, C.A.; Koppler, K.; Vogt, H.; Papadopoulos, N.T. Effects of photoperiod and relative humidity on diapause termination and post-winter development of Rhagoletis cerasi pupae. Bull. Entomol. Res. 2020, 110, 588–596. [Google Scholar] [CrossRef]
- Zhai, Y.; Dong, X.; Gao, H.; Chen, H.; Yang, P.; Li, P.; Yin, Z.; Zheng, L.; Yu, Y. Quantitative Proteomic and Transcriptomic Analyses of Metabolic Regulation of Adult Reproductive Diapause in Drosophila suzukii (Diptera: Drosophilidae) Females. Front. Physiol. 2019, 10, 344. [Google Scholar] [CrossRef]
- Baker, D.A.; Russell, S. Gene expression during Drosophila melanogaster egg development before and after reproductive diapause. BMC Genom. 2009, 10, 242. [Google Scholar] [CrossRef]
- Podrabsky, J.E.; Hand, S.C. Physiological strategies during animal diapause: Lessons from brine shrimp and annual killifish. J. Exp. Biol. 2015, 218, 1897–1906. [Google Scholar] [CrossRef]
- Ragland, G.J.; Egan, S.P.; Feder, J.L.; Berlocher, S.H.; Hahn, D.A. Developmental trajectories of gene expression reveal candidates for diapause termination: A key life-history transition in the apple maggot fly Rhagoletis pomonella. J. Exp. Biol. 2011, 214, 3948–3959. [Google Scholar] [CrossRef]
- An, J.; Huang, J.; Shao, Y.; Zhang, S.; Wang, B.; Liu, X.; Wu, J.; Williams, P.H. The bumblebees of North China (Apidae, Bombus Latreille). Zootaxa 2014, 3830, 1–89. [Google Scholar] [CrossRef] [Green Version]
- Velthuis, H.H.W.; van Doorn, A. A century of advances in bumblebee domestication and the economic and environmental aspects of its commercialization for pollination. Apidologie 2006, 37, 421–451. [Google Scholar] [CrossRef]
- Kim, B.-G.; Shim, J.-K.; Kim, D.-W.; Kwon, Y.J.; Lee, K.-Y. Tissue-specific variation of heat shock protein gene expression in relation to diapause in the bumblebee Bombus terrestris. Entomol. Res. 2008, 38, 10–16. [Google Scholar] [CrossRef]
- Colinet, H.; Pineau, C.; Com, E. Large scale phosphoprotein profiling to explore Drosophila cold acclimation regulatory mechanisms. Sci. Rep. 2017, 7, 1713. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shui, K.; Ma, S.; Lin, S.; Zhang, Y.; Wen, B.; Deng, W.; Xu, H.; Hu, H.; Guo, A.; et al. Integrated omics in Drosophila uncover a circadian kinome. Nat. Commun. 2020, 11, 2710. [Google Scholar] [CrossRef]
- Han, B.; Fang, Y.; Feng, M.; Hu, H.; Hao, Y.; Ma, C.; Huo, X.; Meng, L.; Zhang, X.; Wu, F.; et al. Brain Membrane Proteome and Phosphoproteome Reveal Molecular Basis Associating with Nursing and Foraging Behaviors of Honeybee Workers. J. Proteome Res. 2017, 16, 3646–3663. [Google Scholar] [CrossRef]
- Gala, A.; Fang, Y.; Woltedji, D.; Zhang, L.; Han, B.; Feng, M.; Li, J. Changes of proteome and phosphoproteome trigger embryo-larva transition of honeybee worker (Apis mellifera ligustica). J. Proteom. 2013, 78, 428–446. [Google Scholar] [CrossRef]
- Han, B.; Fang, Y.; Feng, M.; Lu, X.; Huo, X.; Meng, L.; Wu, B.; Li, J. In-depth phosphoproteomic analysis of royal jelly derived from western and eastern honeybee species. J. Proteome Res. 2014, 13, 5928–5943. [Google Scholar] [CrossRef]
- Resende, V.M.; Vasilj, A.; Santos, K.S.; Palma, M.S.; Shevchenko, A. Proteome and phosphoproteome of Africanized and European honeybee venoms. Proteomics 2013, 13, 2638–2648. [Google Scholar] [CrossRef]
- Feng, M.; Fang, Y.; Han, B.; Zhang, L.; Lu, X.; Li, J. Novel aspects of understanding molecular working mechanisms of salivary glands of worker honeybees (Apis mellifera) investigated by proteomics and phosphoproteomics. J. Proteom. 2013, 87, 1–15. [Google Scholar] [CrossRef]
- Qi, Y.; Fan, P.; Hao, Y.; Han, B.; Fang, Y.; Feng, M.; Cui, Z.; Li, J. Phosphoproteomic Analysis of Protein Phosphorylation Networks in the Hypopharyngeal Gland of Honeybee Workers (Apis mellifera ligustica). J. Proteome Res. 2015, 14, 4647–4661. [Google Scholar] [CrossRef]
- Williams, K.D.; Busto, M.; Suster, M.L.; So, A.K.; Ben-Shahar, Y.; Leevers, S.J.; Sokolowski, M.B. Natural variation in Drosophila melanogaster diapause due to the insulin-regulated PI3-kinase. Proc. Natl. Acad. Sci. USA 2006, 103, 15911–15915. [Google Scholar] [CrossRef] [PubMed]
- Hao, K.; Ullah, H.; Jarwar, A.R.; Nong, X.; Tu, X.; Zhang, Z. Molecular identification and diapause-related functional characterization of a novel dual-specificity kinase gene, MPKL, in Locusta migratoria. FEBS Lett. 2019, 593, 3064–3074. [Google Scholar] [CrossRef]
- Lin, J.L.; Lin, P.L.; Gu, S.H. Phosphorylation of glycogen synthase kinase-3beta in relation to diapause processing in the silkworm, Bombyx mori. J. Insect Physiol. 2009, 55, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Kidokoro, K.; Iwata, K.; Takeda, M.; Fujiwara, Y. Involvement of ERK/MAPK in regulation of diapause intensity in the false melon beetle, Atrachya menetriesi. J. Insect Physiol. 2006, 52, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Shindome, C.; Takeda, M.; Shiomi, K. The roles of ERK and P38 MAPK signaling cascades on embryonic diapause initiation and termination of the silkworm, Bombyx mori. Insect Biochem. Mol. Biol. 2006, 36, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.X.; Xu, W.H. Proteomic and Phosphoproteomic Analysis at Diapause Initiation in the Cotton Bollworm, Helicoverpa armigera. J. Proteome Res. 2010, 9, 5053–5064. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.C.; Pavlides, S.A.; Tammariello, S.P. Proteomic and phosphoproteomic profiling during diapause entrance in the flesh fly, Sarcophaga crassipalpis. J. Insect Physiol. 2011, 57, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Hugo, R.L.E.; Birrell, G.W. Proteomics of Anopheles Vectors of Malaria. Trends Parasitol. 2018, 34, 961–981. [Google Scholar] [CrossRef]
- He, X.; Cao, X.; He, Y.; Bhattarai, K.; Rogers, J.; Hartson, S.; Jiang, H. Hemolymph proteins of Anopheles gambiae larvae infected by Escherichia coli. Dev. Comp. Immunol. 2017, 74, 110–124. [Google Scholar] [CrossRef]
- Casas-Vila, N.; Bluhm, A.; Sayols, S.; Dinges, N.; Dejung, M.; Altenhein, T.; Kappei, D.; Altenhein, B.; Roignant, J.Y.; Butter, F. The developmental proteome of Drosophila melanogaster. Genome Res. 2017, 27, 1273–1285. [Google Scholar] [CrossRef] [Green Version]
- Jia, S.H.; Li, M.W.; Zhou, B.; Liu, W.B.; Zhang, Y.; Miao, X.X.; Zeng, R.; Huang, Y.P. Proteomic analysis of silk gland programmed cell death during metamorphosis of the silkworm Bombyx mori. J. Proteome Res. 2007, 6, 3003–3010. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.Y.; Zhou, X.R.; Tan, Y.; Pang, B.P. Proteomic analysis of adult Galeruca daurica (Coleoptera: Chrysomelidae) at different stages during summer diapause. Comp. Biochem. Physiol. Part D Genom. Proteom. 2019, 29, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Tang, L.C.; Krug, K.; Clark, D.J.; Gritsenko, M.A.; Chen, L.; Clauser, K.R.; Clauss, T.R.; Shah, P.; Gillette, M.A.; et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc. 2018, 13, 1632–1661. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Guillemain, A.; Laouarem, Y.; Cobret, L.; Stefok, D.; Chen, W.; Bloch, S.; Zahaf, A.; Blot, L.; Reverchon, F.; Normand, T.; et al. LINGO family receptors are differentially expressed in the mouse brain and form native multimeric complexes. FASEB J. 2020, 34, 13641–13653. [Google Scholar] [CrossRef]
- Ji, Q.; Guo, S.; Wang, X.; Pang, C.; Zhan, Y.; Chen, Y.; An, H. Recent advances in TMEM16A: Structure, function, and disease. J. Cell Physiol. 2019, 234, 7856–7873. [Google Scholar] [CrossRef]
- Amsalem, E.; Galbraith, D.A.; Cnaani, J.; Teal, P.E.; Grozinger, C.M. Conservation and modification of genetic and physiological toolkits underpinning diapause in bumble bee queens. Mol. Ecol. 2015, 24, 5596–5615. [Google Scholar] [CrossRef]
- Chen, H.; Wu, G.; Zhou, H.; Dai, X.; Steeghs, N.W.F.; Dong, X.; Zheng, L.; Zhai, Y. Hormonal Regulation of Reproductive Diapause That Occurs in the Year-Round Mass Rearing of Bombus terrestris Queens. J. Proteome Res. 2021, 20, 2240–2250. [Google Scholar] [CrossRef]
- Colgan, T.J.; Finlay, S.; Brown, M.J.F.; Carolan, J.C. Mating precedes selective immune priming which is maintained throughout bumblebee queen diapause. BMC Genom. 2019, 20, 959. [Google Scholar] [CrossRef]
- Zhang, Q.; Lu, Y.X.; Xu, W.H. Integrated proteomic and metabolomic analysis of larval brain associated with diapause induction and preparation in the cotton bollworm, Helicoverpa armigera. J. Proteome Res. 2012, 11, 1042–1053. [Google Scholar] [CrossRef]
- Zhao, J.Y.; Zhao, X.T.; Sun, J.T.; Zou, L.F.; Yang, S.X.; Han, X.; Zhu, W.C.; Yin, Q.; Hong, X.Y. Transcriptome and proteome analyses reveal complex mechanisms of reproductive diapause in the two-spotted spider mite, Tetranychus urticae. Insect Mol. Biol. 2017, 26, 215–232. [Google Scholar] [CrossRef] [PubMed]
- Divito, C.B.; Underhill, S.M. Excitatory amino acid transporters: Roles in glutamatergic neurotransmission. Neurochem. Int. 2014, 73, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Denlinger, D.L. High temperature and hexane break pupal diapause in the flesh fly, Sarcophaga crassipalpis, by activating ERK/MAPK. J. Insect Physiol. 2007, 53, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Denlinger, D.L. p38 MAPK is a likely component of the signal transduction pathway triggering rapid cold hardening in the flesh fly Sarcophaga crassipalpis. J. Exp. Biol. 2007, 210, 3295–3300. [Google Scholar] [CrossRef]
- Iwata, K.; Shindome, C.; Kobayashi, Y.; Takeda, M.; Yamashita, O.; Shiomi, K.; Fujiwara, Y. Temperature-dependent activation of ERK/MAPK in yolk cells and its role in embryonic diapause termination in the silkworm Bombyx mori. J. Insect Physiol. 2005, 51, 1306–1312. [Google Scholar] [CrossRef]
- Hand, S.C.; Denlinger, D.L.; Podrabsky, J.E.; Roy, R. Mechanisms of animal diapause: Recent developments from nematodes, crustaceans, insects, and fish. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R1193–R1211. [Google Scholar] [CrossRef]
- Erkut, C.; Kurzchalia, T.V. The C. elegans dauer larva as a paradigm to study metabolic suppression and desiccation tolerance. Planta 2015, 242, 389–396. [Google Scholar] [CrossRef]
- Hao, Y.J.; Zhang, Y.J.; Si, F.L.; Fu, D.Y.; He, Z.B.; Chen, B. Insight into the possible mechanism of the summer diapause of Delia antiqua (Diptera: Anthomyiidae) through digital gene expression analysis. Insect Sci. 2016, 23, 438–451. [Google Scholar] [CrossRef]
- Dong, Y.C.; Chen, Z.Z.; Clarke, A.R.; Niu, C.Y. Changes in Energy Metabolism Trigger Pupal Diapause Transition of Bactrocera minax After 20-Hydroxyecdysone Application. Front. Physiol. 2019, 10, 1288. [Google Scholar] [CrossRef]
- Agrell, I. Pupal diapause caused by vitamin deficiency. Nature 1951, 167, 283–284. [Google Scholar] [CrossRef]
- Romney, A.L.T.; Davis, E.M.; Corona, M.M.; Wagner, J.T.; Podrabsky, J.E. Temperature-dependent vitamin D signaling regulates developmental trajectory associated with diapause in an annual killifish. Proc. Natl. Acad. Sci. USA 2018, 115, 12763–12768. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Han, Y.; Yang, Y.; Zhao, X.; Tang, Y.; Li, X.; Diao, Y.; Xu, B. Transcriptomic analysis of ovarian signaling at the emergence of the embryo from obligate diapause in the American mink (Neovison vison). Anim. Reprod. Sci. 2021, 232, 106823. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Sakamoto, K.; Bayascas, J.R. LKB1-dependent signaling pathways. Annu. Rev. Biochem. 2006, 75, 137–163. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Mäkelä, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef]
- Cheng, H.; Liu, P.; Wang, Z.C.; Zou, L.; Santiago, S.; Garbitt, V.; Gjoerup, O.V.; Iglehart, J.D.; Miron, A.; Richardson, A.L.; et al. SIK1 couples LKB1 to p53-dependent anoikis and suppresses metastasis. Sci. Signal. 2009, 2, ra35. [Google Scholar] [CrossRef]
- Carling, D.; Thornton, C.; Woods, A.; Sanders, M.J. AMP-activated protein kinase: New regulation, new roles? Biochem. J. 2012, 445, 11–27. [Google Scholar] [CrossRef]
- Lutzner, N.; Kalbacher, H.; Krones-Herzig, A.; Rosl, F. FOXO3 is a glucocorticoid receptor target and regulates LKB1 and its own expression based on cellular AMP levels via a positive autoregulatory loop. PLoS ONE 2012, 7, e42166. [Google Scholar] [CrossRef]
- Lutzner, N.; De-Castro Arce, J.; Rosl, F. Gene expression of the tumour suppressor LKB1 is mediated by Sp1, NF-Y and FOXO transcription factors. PLoS ONE 2012, 7, e32590. [Google Scholar] [CrossRef]
- Klotz, L.O.; Sanchez-Ramos, C.; Prieto-Arroyo, I.; Urbanek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wang, Y.; Zhu, W.G. Applications of post-translational modifications of FoxO family proteins in biological functions. J. Mol. Cell Biol. 2011, 3, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Guo, S.; Cichy, S.C.; Unterman, T.G.; Cohen, P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem. 1999, 274, 17179–17183. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Chen, H.; Pullikotil, P.; Quon, M.J. Protein kinase A-alpha directly phosphorylates FoxO1 in vascular endothelial cells to regulate expression of vascular cellular adhesion molecule-1 mRNA. J. Biol. Chem. 2011, 286, 6423–6432. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Woods, Y.L.; Prescott, A.R.; Peggie, M.; Unterman, T.G.; Williams, M.R.; Cohen, P. Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. EMBO J. 2002, 21, 2263–2271. [Google Scholar] [CrossRef]
- Yun, H.; Park, S.; Kim, M.J.; Yang, W.K.; Im, D.U.; Yang, K.R.; Hong, J.; Choe, W.; Kang, I.; Kim, S.S.; et al. AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. FEBS J. 2014, 281, 4421–4438. [Google Scholar] [CrossRef]
- Yuan, Z.; Lehtinen, M.K.; Merlo, P.; Villen, J.; Gygi, S.; Bonni, A. Regulation of neuronal cell death by MST1-FOXO1 signaling. J. Biol. Chem. 2009, 284, 11285–11292. [Google Scholar] [CrossRef]
- Asada, S.; Daitoku, H.; Matsuzaki, H.; Saito, T.; Sudo, T.; Mukai, H.; Iwashita, S.; Kako, K.; Kishi, T.; Kasuya, Y.; et al. Mitogen-activated protein kinases, Erk and p38, phosphorylate and regulate Foxo1. Cell. Signal. 2007, 19, 519–527. [Google Scholar] [CrossRef]
- Saline, M.; Badertscher, L.; Wolter, M.; Lau, R.; Gunnarsson, A.; Jacso, T.; Norris, T.; Ottmann, C.; Snijder, A. AMPK and AKT protein kinases hierarchically phosphorylate the N-terminus of the FOXO1 transcription factor, modulating interactions with 14-3-3 proteins. J. Biol. Chem. 2019, 294, 13106–13116. [Google Scholar] [CrossRef]
- Ma, J.; Chen, T.; Wu, S.; Yang, C.; Bai, M.; Shu, K.; Li, K.; Zhang, G.; Jin, Z.; He, F.; et al. iProX: An integrated proteome resource. Nucleic Acids Res. 2019, 47, D1211–D1217. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Accession | Protein Description | Ratio | p Value | Gene Name | MW (kDa) |
|---|---|---|---|---|---|
| Comparable group PD/D up | |||||
| A0A6P3D6E6 | leucine-rich repeat and immunoglobulin-like domain-containing nogo receptor-interacting protein 2 | 11.493 | 5.98 × 10−5 | LOC100648620 | 103.4 |
| A0A6P3DB00 | CDK-activating kinase assembly factor MAT1 | 8.873 | 1.055 × 10−5 | LOC100642207 | 36.711 |
| A0A6P5HS32 | Transmembrane protease serine | 5.623 | 0.0093336 | LOC110119488 | 65.551 |
| A0A6P3UB32 | putative inorganic phosphate cotransporter | 5.601 | 9.643 × 10−5 | LOC100647822 | 53.886 |
| A0A6P3TTL0 | suppressor APC domain-containing protein 2 | 5.545 | 0.0015442 | LOC100647337 | 51.622 |
| A0A6P5I390 | E3 ubiquitin-protein ligase Rnf220 | 5.301 | 0.0002411 | LOC100643542 | 35.027 |
| A0A6P3DC42 | disheveled-associated activator of morphogenesis 1 | 5.122 | 9.54 × 10−6 | LOC100650848 | 123.68 |
| Comparable group FPD/PD up | |||||
| A0A6P5IBX2 | anoctamin-1 | 0.551 | 0.0077095 | LOC100646632 | 115.18 |
| A0A6P3DGE6 | pancreatic lipase-related protein 2 | 0.99 | 0.8897713 | LOC100645800 | 34.752 |
| A0A6P3U8X6 | phospholipase A2 | 1.159 | 0.2525801 | LOC100651730 | 22.336 |
| A0A6P3TVD7 | cation-independent mannose-6-phosphate receptor | 0.898 | 0.098125 | LOC100651480 | 82.845 |
| A0A6P3DNC4 | serine protease inhibitor 3/4 | 1.356 | 0.0453889 | LOC100652301 | 35.236 |
| A0A6P3DFC5 | nuclear factor related to kappa-B-binding protein | 1.138 | 0.1405269 | LOC100649343 | 168.64 |
| A0A6P5HLS9 | protein toll | 1.775 | 0.0262693 | LOC100651716 | 125.92 |
| A0A6P3U2L5 | Apoptosis-stimulating of p53 | 1.607 | 0.02605 | LOC100646232 | 139.27 |
| A0A6P3TPE5 | venom protease | 1.551 | 0.0013554 | LOC100642484 | 32.966 |
| A0A6P3TWR2 | TNF receptor-associated factor 5 | 2.023 | 0.0232261 | LOC105665677 | 55.742 |
| A0A6P3UKF1 | venom acid phosphatase Acph-1 | 1.208 | 0.1376568 | LOC100647178 | 48.343 |
| A0A6P5I2Z1 | major royal jelly protein 1 | 2.115 | 0.0755501 | LOC100648898 | 41.083 |
| A0A6P3UGV4 | basement membrane-specific heparan sulfate proteoglycan core protein | 0.067 | 0.0001312 | LOC100651429 | 468.9 |
| A0A6P5HLF4 | CLIP domain-containing serine protease | 1.181 | 0.0913721 | LOC100652157 | 82.898 |
| A0A6P3TNQ5 | alpha-glucosidase | 1.321 | 0.0721932 | LOC100643608 | 65.637 |
| A0A6P3DF18 | facilitated trehalose transporter Tret1-lik | 0.811 | 0.1431761 | LOC100644978 | 76.971 |
| Protein Accession | Position | Amino Acid | Protein Description | Ratio | p Value | Gene Name |
|---|---|---|---|---|---|---|
| Comparable group PD/D up | ||||||
| A0A6P3U5V0 | 91 | S | RNA polymerase-associated protein LEO1 | 9.982 | 4.011 × 10−5 | LOC100652289 |
| A0A6P3DA37 | 471 | S | dolichyl-diphosphooligosaccharide--protein glycosyltransferase subunit STT3B | 9.306 | 0.0002187 | LOC100647181 |
| A0A6P5IE28 | 166 | S | catenin alpha | 8.092 | 0.0001623 | LOC100648462 |
| A0A6P5I410 | 505 | T | splicing factor 3B subunit 2 | 8.01 | 0.0005079 | LOC100643117 |
| A0A6P3TWI1 | 591 | S | DDB1- and CUL4-associated factor 8 | 7.232 | 0.0001319 | LOC100644051 |
| A0A6P3U5V0 | 119 | S | RNA polymerase-associated protein LEO1 | 6.862 | 0.0001676 | LOC100652289 |
| A0A6P3DB01 | 580 | S | nucleolin | 6.187 | 3.099 × 10−6 | LOC100644519 |
| A0A6P3UJ38 | 26 | S | GD14428 | 6.181 | 3.796 × 10−5 | LOC100643604 |
| A0A6P3UFI4 | 228 | T | microtubule-actin cross-linking factor 1 | 5.57 | 6.545 × 10−5 | LOC100647948 |
| A0A6P3UAY9 | 147 | S | solute carrier family 25 member 38 | 5.54 | 4.793 × 10−5 | LOC100649391 |
| A0A6P3UJ22 | 273 | S | 60S ribosomal protein L5 | 5.503 | 0.006308 | LOC100642874 |
| A0A6P5HLM2 | 926 | S | trichohyalin | 5.485 | 9.834 × 10−5 | LOC100646259 |
| A0A6P3U0R3 | 2381 | S | golgin subfamily A member 4 | 5.469 | 0.0001002 | LOC105665975 |
| A0A6P5I2T4 | 771 | S | tau-tubulin kinase homolog Asator | 5.344 | 0.0068525 | LOC100646154 |
| A0A6P5HGT7 | 692 | S | serine/threonine-protein kinase MARK2 isoform X11 | 5.248 | 0.000345 | LOC100645231 |
| A0A6P5HR14 | 1421 | T | myosin heavy chain, muscle | 5.237 | 0.0001658 | LOC100647343 |
| A0A6P3D774 | 1536 | T | lysine-specific demethylase lid | 5.103 | 0.0060939 | LOC100648929 |
| A0A6P5HR09 | 5303 | T | titin | 5.094 | 0.0011792 | LOC100643650 |
| A0A6P3D7R3 | 197 | S | 60S ribosomal protein L15 | 5.046 | 0.0008772 | LOC100648934 |
| Comparable group FPD/PD up | ||||||
| A0A6P3TLJ9 | 504 | T | vitellogenin | 10.046 | 0.0048077 | LOC100650436 |
| A0A6P3U9T5 | 23 | S | sugar transporter ERD6-like 8 | 8.168 | 0.0001362 | LOC100644377 |
| A0A6P3U996 | 454 | S | serine/arginine repetitive matrix protein 2 | 7.991 | 0.0094707 | LOC100642774 |
| A0A6P3UGK1 | 319 | S | jerky protein homolog-like | 7.445 | 0.0183674 | LOC105666848 |
| A0A6P3U6P7 | 1604 | S | TBC1 domain family member 30 | 5.581 | 0.1101188 | LOC100645056 |
| A0A6P3DHB6 | 22 | T | synaptic vesicle membrane protein VAT-1 homolog-like | 5.525 | 0.0003307 | LOC100650297 |
| A0A6P3DFL1 | 81 | S | Katnb1_0 protein | 5.434 | 0.0002241 | LOC100645647 |
| A0A6P3DF18 | 681 | S | facilitated trehalose transporter Tret1-like | 5.329 | 0.0006047 | LOC100644978 |
| A0A6P3UBL9 | 551 | S | Heparanase | 5.221 | 0.0251118 | LOC100646845 |
| Group | Motif Logo | Motif | Motif Score | Fold Change | Putative Protein Kinases |
|---|---|---|---|---|---|
| PD vs. D up |  | Xxxxxx_S_DxExxx | 32.00 | 11.0 | novel |
| PD vs. D down |  | Xxxxxx_T_PPxxxx | 27.44 | 38.2 | cyclin dependent kinase 7 substrate motif |
| FPD vs. PD up |  | Xxxxxx_T_PPxxxx | 24.2 | 39.9 | cyclin dependent kinase 1 |
| FPD vs. PD down |  | xxxKxx_S_Pxxxxx | 25.46 | 14.8 | novel |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Wang, R.; Su, L.; Zhao, S.; Dai, X.; Chen, H.; Wu, G.; Zhou, H.; Zheng, L.; Zhai, Y. Integrative Proteomic and Phosphoproteomic Analyses Revealed Complex Mechanisms Underlying Reproductive Diapause in Bombus terrestris Queens. Insects 2022, 13, 862. https://doi.org/10.3390/insects13100862
Liu Y, Wang R, Su L, Zhao S, Dai X, Chen H, Wu G, Zhou H, Zheng L, Zhai Y. Integrative Proteomic and Phosphoproteomic Analyses Revealed Complex Mechanisms Underlying Reproductive Diapause in Bombus terrestris Queens. Insects. 2022; 13(10):862. https://doi.org/10.3390/insects13100862
Chicago/Turabian StyleLiu, Yan, Ruijuan Wang, Long Su, Shan Zhao, Xiaoyan Dai, Hao Chen, Guang’an Wu, Hao Zhou, Li Zheng, and Yifan Zhai. 2022. "Integrative Proteomic and Phosphoproteomic Analyses Revealed Complex Mechanisms Underlying Reproductive Diapause in Bombus terrestris Queens" Insects 13, no. 10: 862. https://doi.org/10.3390/insects13100862