
Feed and Host Genetics Drive Microbiome Diversity with Resultant Consequences for Production Traits in Mass-Reared Black Soldier Fly (Hermetia illucens) Larvae


 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Larval Rearing, Sample Collection, and Gut Excision
2.2. DNA Extraction and NGS Library Preparation
2.3. Read Processing, Data Filtering, and Normalisation
2.4. Gut Microbiome Diversity and Differentiation Analysis
2.5. Assessment of Core and Enriched Bacterial Genera
2.6. Relationships between Phenotype and Microbiome Data
2.7. Inference of Gut Functional Profiles
3. Results
3.1. Read and OTU Processing
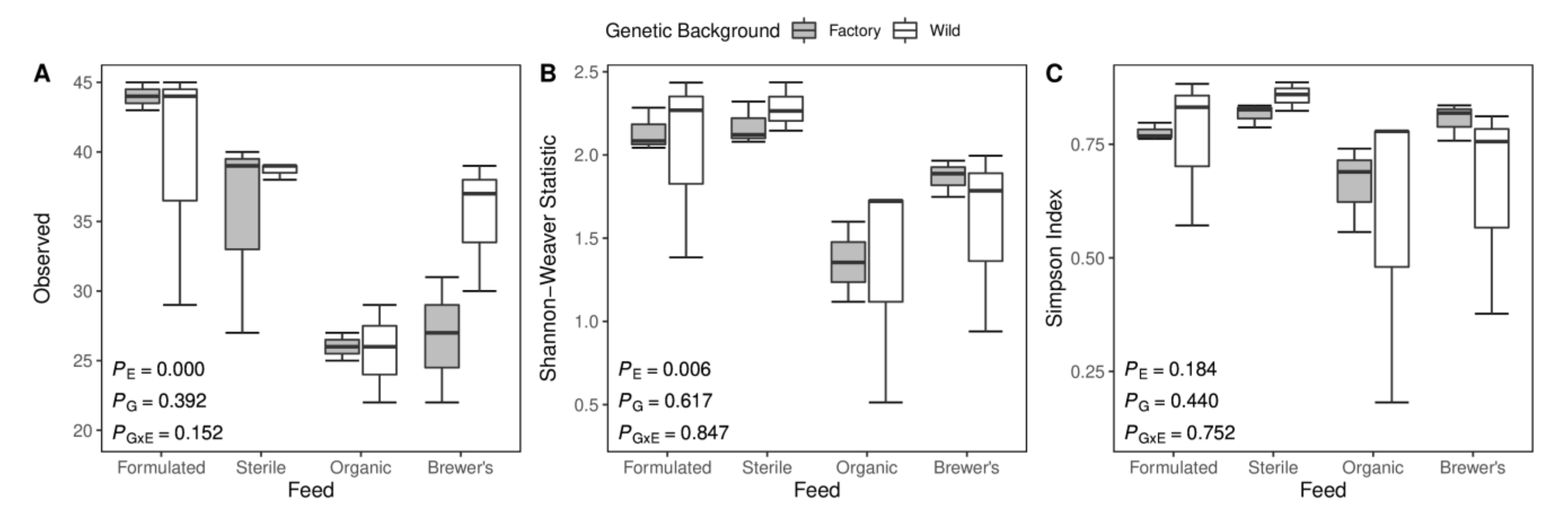
3.2. Measures of Microbial Alpha Diversity
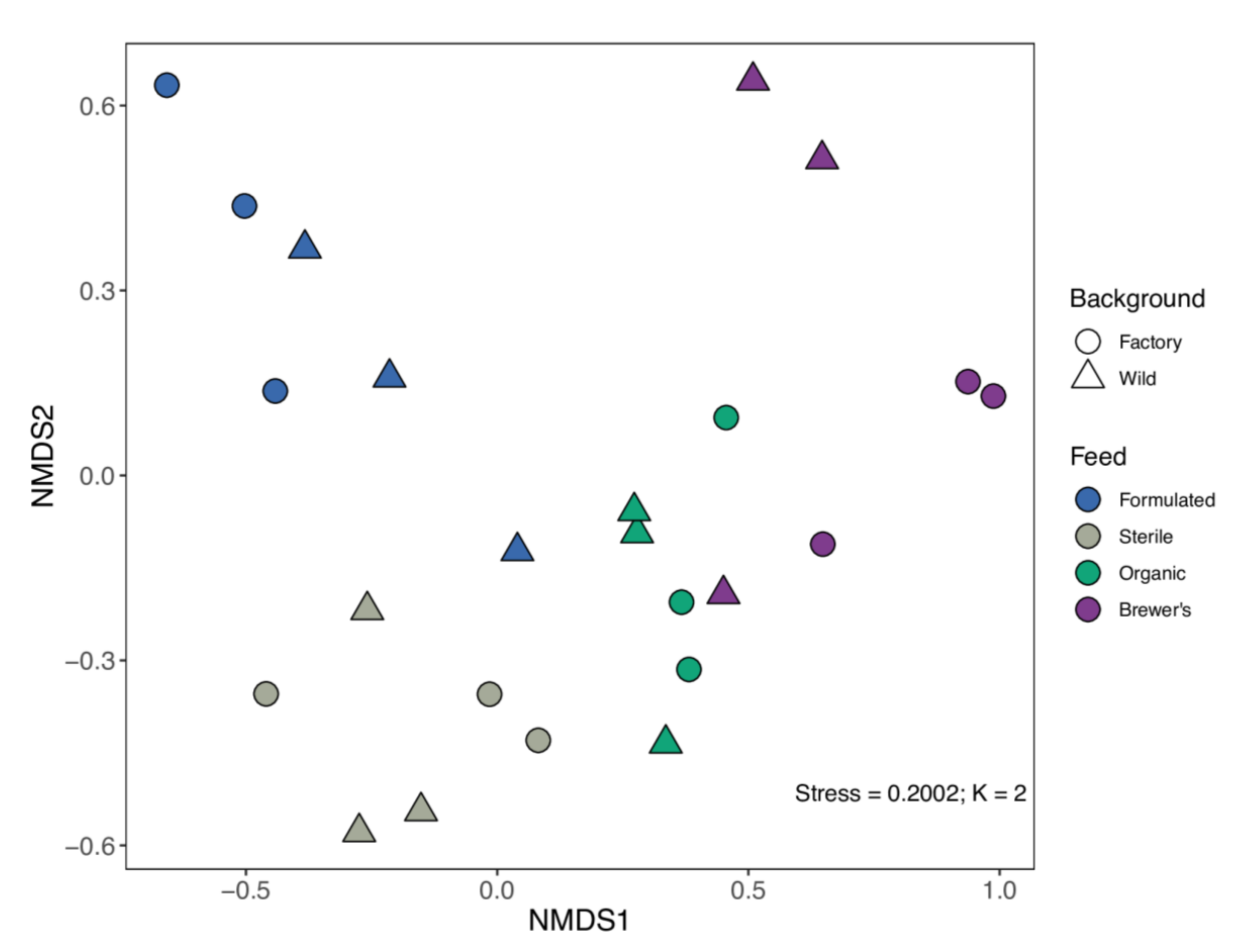
3.3. Microbiome Structure and Differentiation
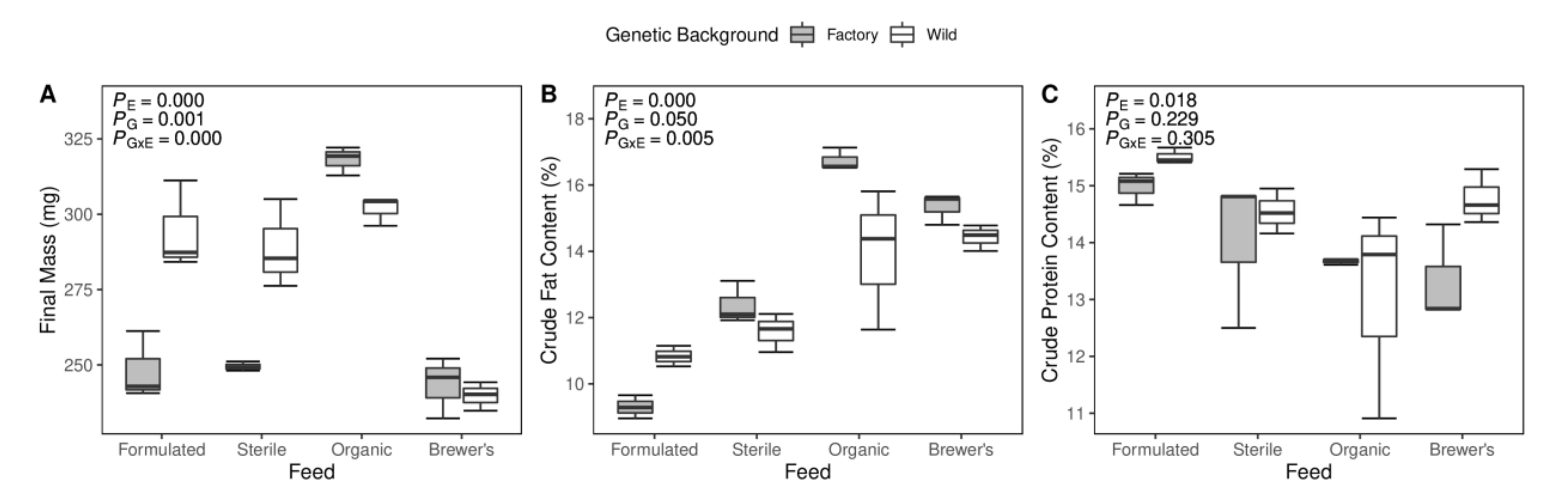
3.4. Production Trait Measures and Relationships to Microbiome Structure
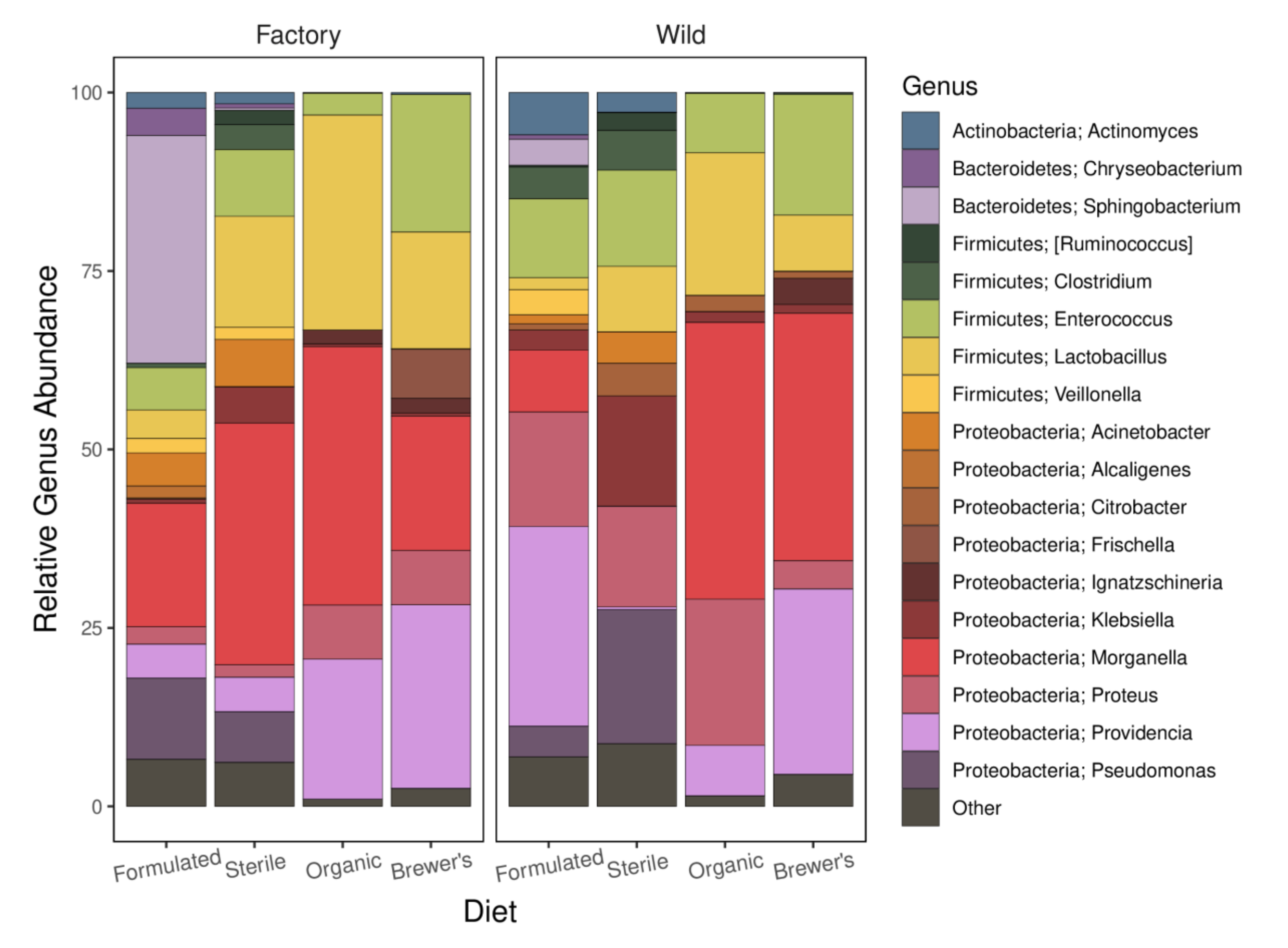
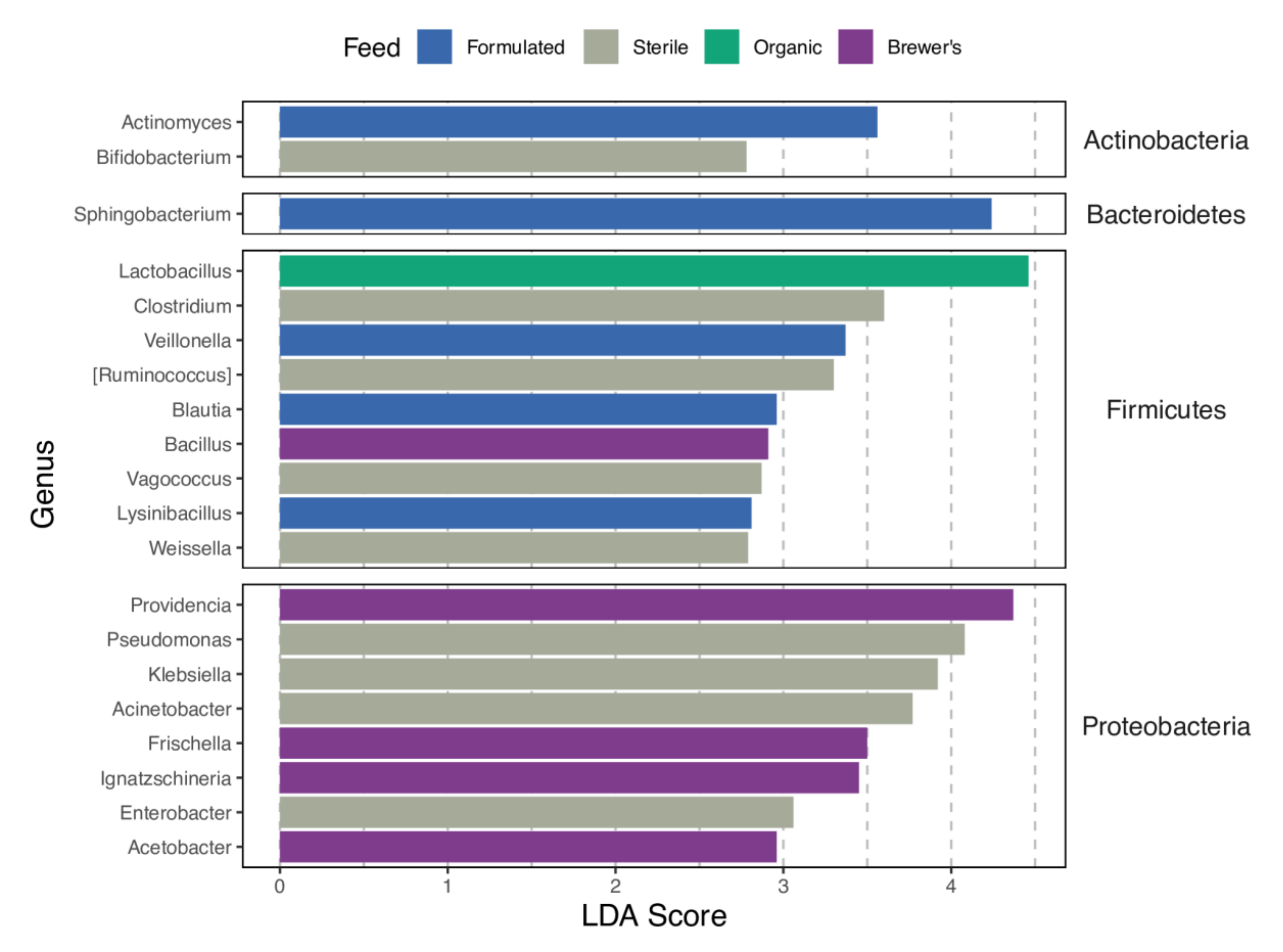
3.5. Taxonomic Profiles, Core Genera, and Patterns of Enrichment
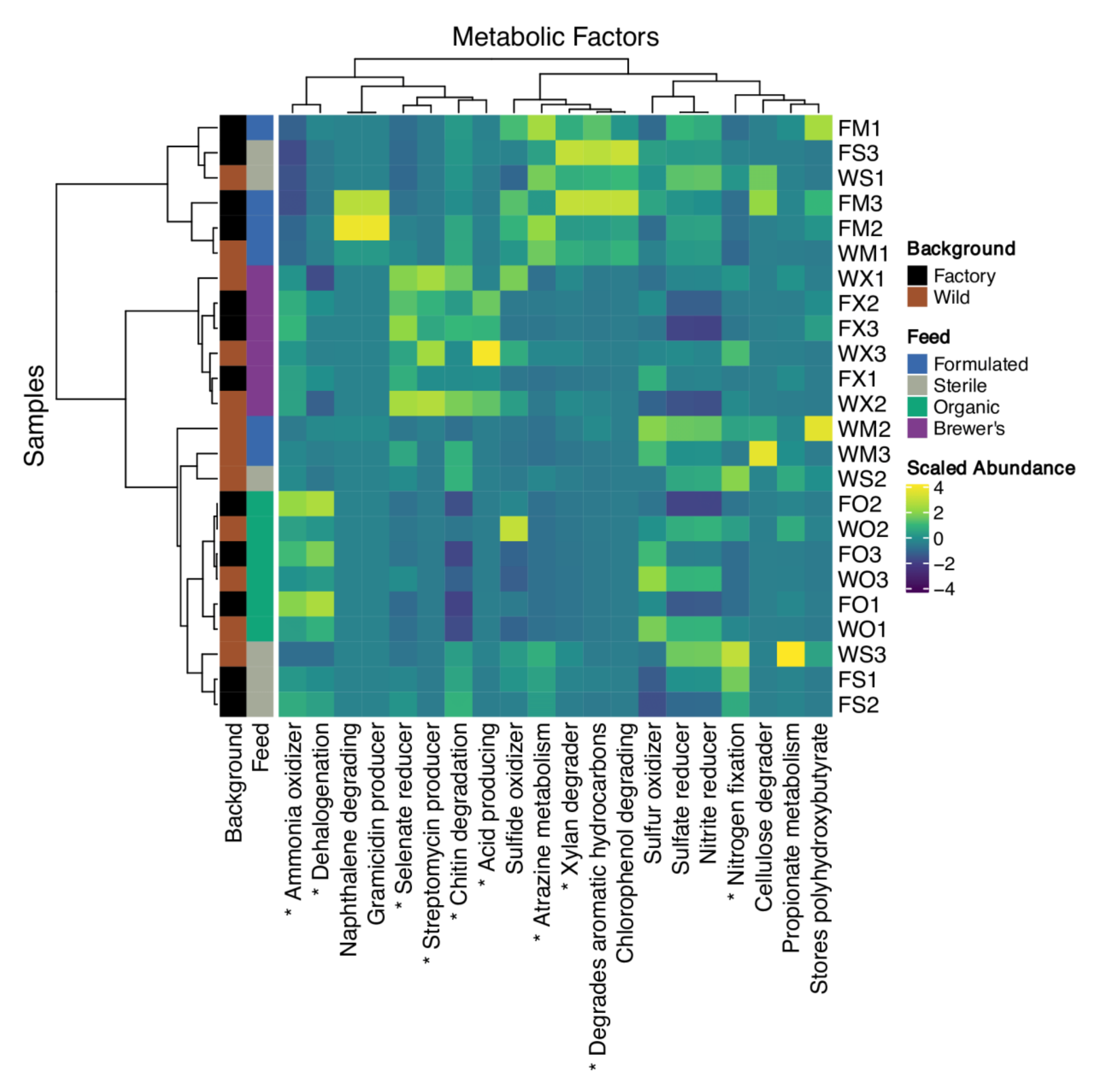
3.6. Inference of Gut Functional Profiles
4. Discussion
4.1. Gut Microbiome Diversity Is Driven by Feed and Host Genetics
4.2. Overarching Gut Microbiome Structure Is Related to Larval Fat Content
4.3. The Implications of Taxonomic and Metabolic Gut Microbiome Profiles
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Godfray, H.C.J.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food security: The challenge of feeding 9 billion people. Science 2010, 327, 812–818. [Google Scholar] [CrossRef] [Green Version]
- Alexander, P.; Brown, C.; Arneth, A.; Finnigan, J.; Moran, D.; Rounsevell, M.D.A. Losses, inefficiencies and waste in the global food system. Agric. Syst. 2017, 153, 190–200. [Google Scholar] [CrossRef]
- Allegretti, G.; Talamini, E.; Schmidt, V.; Bogorni, P.C.; Ortega, E. Insect as feed: An energy assessment of insect meal as a sustainable protein source for the Brazilian poultry industry. J. Clean. Prod. 2018, 171, 403–412. [Google Scholar] [CrossRef]
- Aiking, H. Future protein supply. Trends Food Sci. Technol. 2011, 22, 112–120. [Google Scholar] [CrossRef]
- Ahmed, N.; Thompson, S.; Glaser, M. Global aquaculture productivity, environmental sustainability, and climate change adaptability. Environ. Manag. 2019, 63, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Rumpold, B.A.; Schlüter, O.K. Potential and challenges of insects as an innovative source for food and feed production. Innov. Food Sci. Emerg. Technol. 2013, 17, 1–11. [Google Scholar] [CrossRef]
- Van Huis, A.; Van Itterbeeck, J.; Mertens, E.; Halloran, A.; Muir, G.; Vantomme, P. Edible Insects: Future Prospects for Food and Feed Security; FAO: Rome, Italy, 2013. [Google Scholar]
- Van Huis, A. Potential of insects as food and feed in assuring food security. Ann. Rev. Entomol. 2013, 58, 563–583. [Google Scholar] [CrossRef]
- Barragan-Fonseca, K.B.; Dicke, M.; van Loon, J.J.A. Nutritional value of the black soldier fly (Hermetia illucens L.) and its suitability as animal feed—A review. J. Insects Food Feed 2017, 3, 105–120. [Google Scholar] [CrossRef]
- Ur Rehman, K.; Rehman, A.; Cai, M.; Zheng, L.; Xiao, X.; Somroo, A.A.; Wang, H.; Li, W.; Yu, Z.; Zhang, J. Conversion of mixtures of dairy manure and soybean curd residue by black soldier fly larvae (Hermetia illucens L.). J. Clean. Prod. 2017, 154, 366–373. [Google Scholar] [CrossRef]
- Li, W.; Li, Q.; Zheng, L.; Wang, Y.; Zhang, J.; Yu, Z.; Zhang, Y. Potential biodiesel and biogas production from corncob by anaerobic fermentation and black soldier fly. Bioresour. Technol. 2015, 194, 276–282. [Google Scholar] [CrossRef]
- Caligiani, A.; Marseglia, A.; Leni, G.; Baldassarre, S.; Maistrello, L.; Dossena, A.; Sforza, S. Composition of black soldier fly prepupae and systematic approaches for extraction and fractionation of proteins, lipids and chitin. Food Res. Int. 2018, 105, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.X.; Tomberlin, J.K.; Vanlaerhoven, S. Ability of black soldier fly (Diptera: Stratiomyidae) larvae to recycle food waste. Environ. Entomol. 2015, 44, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Hadj Saadoun, J.; Montevecchi, G.; Zanasi, L.; Bortolini, S.; Macavei, L.I.; Masino, F.; Maistrello, L.; Antonelli, A. Lipid profile and growth of black soldier flies (Hermetia illucens, Stratiomyidae) reared on by-products from different food chains. J. Sci. Food Agric. 2020, 100, 3648–3657. [Google Scholar] [CrossRef]
- Liu, C.; Wang, C.; Yao, H. Comprehensive resource utilization of waste using the black soldier fly (Hermetia illucens (L.)) (Diptera: Stratiomyidae). Animals 2019, 9, 349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Smet, J.; Wynants, E.; Cos, P.; Van Campenhout, L. Microbial community dynamics during rearing of black soldier fly larvae (Hermetia illucens) and impact on exploitation potential. Appl. Environ. Microbiol. 2018, 84, e02722-17. [Google Scholar] [CrossRef] [Green Version]
- Tomberlin, J.K.; Sheppard, D.C.; Joyce, J.A. Selected life-history traits of black soldier flies (Diptera: Stratiomyidae) reared on three artificial diets. Ann. Entomol. Soc. Am. 2002, 95, 379–386. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, L.; He, J.; Tomberlin, J.K.; Li, J.; Lei, C.; Sun, M.; Liu, Z.; Yu, Z. An artificial light source influences mating and oviposition of black soldier flies, Hermetia illucens. J. Insect Sci. 2010, 10, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, L.A.; VanLaerhoven, S.L.; Tomberlin, J.K. Relative humidity effects on the life history of Hermetia illucens (Diptera: Stratiomyidae). Environ. Entomol. 2012, 41, 971–978. [Google Scholar] [CrossRef] [Green Version]
- Chia, S.Y.; Tanga, C.M.; Khamis, F.M.; Mohamed, S.A.; Salifu, D.; Sevgan, S.; Fiaboe, K.K.M.; Niassy, S.; van Loon, J.J.A.; Dicke, M.; et al. Threshold temperatures and thermal requirements of black soldier fly Hermetia illucens: Implications for mass production. PLoS ONE 2018, 13, e0206097. [Google Scholar] [CrossRef] [Green Version]
- Rhode, C.; Badenhorst, R.; Hull, K.L.; Greenwood, M.P.; Bester-van der Merwe, A.E.; Andere, A.A.; Picard, C.J.; Richards, C. Genetic and phenotypic consequences of early domestication in black soldier flies (Hermetia illucens). Anim. Genet. 2020, 51, 752–762. [Google Scholar] [CrossRef]
- Zhan, S.; Fang, G.; Cai, M.; Kou, Z.; Xu, J.; Cao, Y.; Bai, L.; Zhang, Y.; Jiang, Y. Genomic landscape and genetic manipulation of the black soldier fly Hermetia illucens, a natural waste recycler. Cell Res. 2019, 30, 50–60. [Google Scholar] [CrossRef]
- Hayes, B.J.; Lewin, H.A.; Goddard, M.E. The future of livestock breeding: Genomic selection for efficiency, reduced emissions intensity, and adaptation. Trends Genet. 2013, 29, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Georges, M.; Charlier, C.; Hayes, B. Harnessing genomic information for livestock improvement. Nat. Rev. Genet. 2019, 20, 135–156. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, T.; Picard, C.J. Genetic and genomic selection in insects as food and feed. J. Insects Food Feed 2021, 7, 661–682. [Google Scholar] [CrossRef]
- Engel, P.; Moran, N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef]
- Douglas, A.E. The microbial dimension in insect nutritional ecology. Funct. Ecol. 2009, 23, 38–47. [Google Scholar] [CrossRef]
- Benson, A.K. The gut microbiome—An emerging complex trait. Nat. Publ. Gr. 2016, 48, 1301–1302. [Google Scholar] [CrossRef] [PubMed]
- Blekhman, R.; Goodrich, J.K.; Huang, K.; Sun, Q.; Bukowski, R.; Bell, J.T.; Spector, T.D.; Keinan, A.; Ley, R.E.; Gevers, D.; et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015, 16, 191. [Google Scholar] [CrossRef] [Green Version]
- Benson, A.K.; Kelly, S.A.; Legge, R.; Ma, F.; Low, S.J.; Kim, J.; Zhang, M.; Oh, P.L.; Nehrenberg, D.; Hua, K.; et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 2010, 107, 18933–18938. [Google Scholar] [CrossRef] [Green Version]
- Kreznar, J.H.; Keller, M.P.; Traeger, L.L.; Rabaglia, M.E.; Schueler, K.L.; Stapleton, D.S.; Zhao, W.; Vivas, E.I.; Yandell, B.S.; Broman, A.T.; et al. Host genotype and gut microbiome modulate insulin secretion and diet-induced metabolic phenotypes. Cell Rep. 2017, 18, 1739–1750. [Google Scholar] [CrossRef]
- Gold, M.; Binggeli, M.; Kurt, F.; de Wouters, T.; Reichlin, M.; Zurbrügg, C.; Mathys, A.; Kreuzer, M. Novel experimental methods for the investigation of Hermetia illucens (Diptera: Stratiomyidae) larvae. J. Insect Sci. 2020, 20, 21. [Google Scholar] [CrossRef]
- Somroo, A.A.; ur Rehman, K.; Zheng, L.; Cai, M.; Xiao, X.; Hu, S.; Mathys, A.; Gold, M.; Yu, Z.; Zhang, J. Influence of Lactobacillus buchneri on soybean curd residue co-conversion by black soldier fly larvae (Hermetia illucens) for food and feedstock production. Waste Manag. 2019, 86, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Mazza, L.; Xiao, X.; ur Rehman, K.; Cai, M.; Zhang, D.; Fasulo, S.; Tomberlin, J.K.; Zheng, L.; Soomro, A.A.; Yu, Z.; et al. Management of chicken manure using black soldier fly (Diptera: Stratiomyidae) larvae assisted by companion bacteria. Waste Manag. 2020, 102, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Bruno, D.; Bonelli, M.; De Filippis, F.; Di Lelio, I.; Tettamanti, G.; Casartelli, M.; Ercolini, D.; Caccia, S. The intestinal microbiota of Hermetia illucens larvae is affected by diet and shows a diverse composition in the different midgut regions. Appl. Environ. Microbiol. 2019, 85, e01864-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynants, E.; Frooninckx, L.; Crauwels, S.; Verreth, C.; De Smet, J.; Sandrock, C.; Wohlfahrt, J.; Van Schelt, J.; Depraetere, S.; Lievens, B.; et al. Assessing the microbiota of black soldier fly larvae (Hermetia illucens) reared on organic waste streams on four different locations at laboratory and large scale. Microb. Ecol. 2019, 77, 913–930. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Jin, W.; Tao, X.; Zhang, Q.; Zhu, J.; Feng, S.; Xu, X.; Li, H.; Wang, Z.; Zhang, Z. Black soldier fly larvae (Hermetia illucens) strengthen the metabolic function of food waste biodegradation by gut microbiome. Microb. Biotechnol. 2019, 12, 528–543. [Google Scholar] [CrossRef] [Green Version]
- Khamis, F.M.; Ombura, F.L.O.; Akutse, K.S.; Subramanian, S.; Mohamed, S.A.; Fiaboe, K.K.M.; Saijuntha, W.; Van Loon, J.J.A.; Dicke, M.; Dubois, T.; et al. Insights in the global genetics and gut microbiome of black soldier fly, Hermetia illucens: Implications for animal feed safety control. Front. Microbiol. 2020, 11, 1538. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.A.; Woodley, N.E.; Hauser, M. The historical spread of the black soldier fly, Hermetia illucens (L.) (Diptera, Stratiomyidae, Hermetiinae), and its establishment in Canada. J. Entomol. Soc. Ont. 2015, 146, 51–54. [Google Scholar] [CrossRef] [Green Version]
- AOAC. AOAC Official Method 990.03, Protein (Crude) in Animal Feed, Combustion Method; AOAC International: Gaithersburg, MD, USA, 2006. [Google Scholar]
- Müller, J. Dumas or Kjeldahl for Reference Analysis? FOSS: Hilleroed, Denmark, 2017. [Google Scholar]
- Finke, M.D. Estimate of chitin in raw whole insects. Zoo Biol. 2007, 26, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Am, A.A.P. Rapid Determination of Oil/Fat Utilizing High Temperature Solvent Extraction; American Oil Chemists Society: Urbana, IL, USA, 2005. [Google Scholar]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Simpson, E.H. Measurment of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Levene, H. Levene test for equality of variances. In Contributions to Probability and Statistics: Essays in Honor of Harold Hotelling; Stanford University Press: Palo Alto, CA, USA, 1960; pp. 278–292. [Google Scholar]
- Shapiro, S.S.; Wilk, M.B. An analysis of variance test for normality (complete samples). Biometrika 1965, 52, 591–611. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Anderson, M.J. Distance-based tests for homogeneity of multivariate dispersions. Biometrics 2006, 62, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Austral. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Hervé, M. RVAideMemoire: Testing and Plotting Procedures for Biostatistics. 2018. Available online: https://CRAN.R-project.org/package=RVAideMemoire (accessed on 8 December 2020).
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, D.; Xia, J.; Liu, Y.; Zhou, Y.; Guo, A.C.; Cruz, J.A.; Sinelnikov, I.; Budwill, K.; Nesbo, C.L.; Wishart, D.S. METAGENassist: A comprehensive web server for comparative metagenomics. Nucleic Acids Res. 2012, 40, W88–W95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, J.; Liu, Y.; Liang, Y.; Zhou, Y.; Wilson, M.; Dennis, J.J.; Stothard, P.; Van Domselaar, G.; Wishart, D.S. BacMap: An up-to-date electronic atlas of annotated bacterial genomes. Nucleic Acids Res. 2012, 40, D599–D604. [Google Scholar] [CrossRef] [Green Version]
- Pagani, I.; Liolios, K.; Jansson, J.; Chen, I.-M.A.; Smirnova, T.; Nosrat, B.; Markowitz, V.M.; Kyrpides, N.C. The Genomes OnLine Database (GOLD) v.4: Status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2012, 40, D571–D579. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, W.; Schleifer, K.-H.; Whitman, W.B. Revised road map to the phylum Firmicutes. In Bergey’s Manual of Systematic Bacteriology; De Vos, P., Garrity, G.M., Jones, D., Krieg, N.R., Ludwig, W., Rainey, F.A., Schleifer, K.-H., Whitman, W.B., Eds.; Springer: New York, NY, USA, 2009; Volume 3, pp. 1–13. [Google Scholar]
- Leprêtre, A.; Mouillot, D. A comparison of species diversity estimators. Popul. Ecol. 1999, 41, 203–215. [Google Scholar] [CrossRef]
- Bjerre, R.D.; Hugerth, L.W.; Boulund, F.; Seifert, M.; Johansen, J.D.; Engstrand, L. Effects of sampling strategy and DNA extraction on human skin microbiome investigations. Sci. Rep. 2019, 9, 17287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willner, D.; Daly, J.; Whiley, D.; Grimwood, K.; Wainwright, C.E.; Hugenholtz, P. Comparison of DNA extraction methods for microbial community profiling with an application to pediatric bronchoalveolar lavage samples. PLoS ONE 2012, 7, e34605. [Google Scholar] [CrossRef] [Green Version]
- Lim, M.Y.; Song, E.-J.; Kim, S.H.; Lee, J.; Nam, Y.-D. Comparison of DNA extraction methods for human gut microbial community profiling. Syst. Appl. Microbiol. 2018, 41, 151–157. [Google Scholar] [CrossRef]
- Gill, C.; van de Wijgert, J.H.H.M.; Blow, F.; Darby, A.C. Evaluation of lysis methods for the extraction of bacterial DNA for analysis of the vaginal microbiota. PLoS ONE 2016, 11, e0163148. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.; Park, S.; Choi, J.; Jeong, G.; Lee, S.-B.; Choi, Y.; Lee, S.-J. The intestinal bacterial community in the food waste-reducing larvae of Hermetia illucens. Curr. Microbiol. 2011, 62, 1390–1399. [Google Scholar] [CrossRef]
- Klammsteiner, T.; Walter, A.; Bogataj, T.; Heussler, C.D.; Stres, B.; Steiner, F.M.; Schlick-Steiner, B.C.; Arthofer, W.; Insam, H. The core gut microbiome of black soldier fly (Hermetia illucens) larvae raised on low-bioburden diets. Front. Microbiol. 2020, 11, 993. [Google Scholar] [CrossRef]
- Pérez-Cobas, A.E.; Maiques, E.; Angelova, A.; Carrasco, P.; Moya, A.; Latorre, A. Diet shapes the gut microbiota of the omnivorous cockroach Blattella germanica. FEMS Microbiol. Ecol. 2015, 91, fiv022. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, E.A.; Velazquez, K.T.; Herbert, K.M. Influence of high-fat diet on gut microbiota: A driving force for chronic disease risk. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.; Yang, Y.; Luo, Z.; Guan, L.; Zhu, W. The colonic microbiome and epithelial transcriptome are altered in rats fed a high-protein diet compared with a normal-protein diet. J. Nutr. 2016, 146, 474–483. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Lauber, C.L.; Czarnecki-Maulden, G.; Pan, Y.; Hannah, S.S. Effects of the dietary protein and carbohydrate ratio on gut microbiomes in dogs of different body conditions. mBio 2017, 8, e01703-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Rasmussen, T.S.; Rasmussen, M.L.; Li, J.; Henríquez Olguín, C.; Kot, W.; Nielsen, D.S.; Jensen, T.E. The gut microbiome on a periodized low-protein diet is associated with improved metabolic health. Front. Microbiol. 2019, 10, 709. [Google Scholar] [CrossRef] [Green Version]
- Erkosar, B.; Yashiro, E.; Zajitschek, F.; Friberg, U.; Maklakov, A.A.; van der Meer, J.R.; Kawecki, T.J. Host diet mediates a negative relationship between abundance and diversity of Drosophila gut microbiota. Ecol. Evol. 2018, 8, 9491–9502. [Google Scholar] [CrossRef]
- Spor, A.; Koren, O.; Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Nishida, A.H.; Ochman, H. Rates of gut microbiome divergence in mammals. Mol. Ecol. 2018, 27, 1884–1897. [Google Scholar] [CrossRef]
- Colman, D.R.; Toolson, E.C.; Takacs-Vesbach, C.D. Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 2012, 21, 5124–5137. [Google Scholar] [CrossRef]
- Turpin, W.; Espin-Garcia, O.; Xu, W.; Silverberg, M.S.; Kevans, D.; Smith, M.I.; Guttman, D.S.; Griffiths, A.; Panaccione, R.; Otley, A.; et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat. Genet. 2016, 48, 1413–1417. [Google Scholar] [CrossRef]
- Org, E.; Parks, B.W.; Joo, J.W.J.; Emert, B.; Schwartzman, W.; Kang, E.Y.; Mehrabian, M.; Pan, C.; Knight, R.; Gunsalus, R.; et al. Genetic and environmental control of host-gut microbiota interactions. Genome Res. 2015, 25, 1558–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, E.R. Elucidating the role of the host genome in shaping microbiome composition. Gut Microbes 2016, 7, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Camarinha-Silva, A.; Maushammer, M.; Wellmann, R.; Vital, M.; Preuss, S.; Bennewitz, J. Host genome influence on gut microbial composition and microbial prediction of complex traits in pigs. Genetics 2017, 206, 1637–1644. [Google Scholar] [CrossRef]
- Grieneisen, L.; Dasari, M.; Gould, T.J.; Björk, J.R.; Grenier, J.; Yotova, V.; Jansen, D.; Gottel, N.; Gordon, J.B.; Learn, N.H.; et al. Gut microbiome heritability is nearly universal but environmentally contingent. Science 2021, 373, 181–186. [Google Scholar] [CrossRef]
- Bonder, M.J.; Kurilshikov, A.; Tigchelaar, E.F.; Mujagic, Z.; Imhann, F.; Vila, A.V.; Deelen, P.; Vatanen, T.; Schirmer, M.; Smeekens, S.P.; et al. The effect of host genetics on the gut microbiome. Nat. Genet. 2016, 48, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Davenport, E.R.; Waters, J.L.; Clark, A.G.; Ley, R.E. Cross-species comparisons of host genetic associations with the microbiome. Science 2016, 352, 532–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Kalyan, S.; Steck, N.; Turner, L.M.; Harr, B.; Künzel, S.; Vallier, M.; Häsler, R.; Franke, A.; Oberg, H.-H.; et al. Analysis of intestinal microbiota in hybrid house mice reveals evolutionary divergence in a vertebrate hologenome. Nat. Commun. 2015, 6, 6440. [Google Scholar] [CrossRef] [Green Version]
- Benson, A.K. Host genetic architecture and the landscape of microbiome composition: Humans weigh in. Genome Biol. 2015, 16, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leamy, L.J.; Kelly, S.A.; Nietfeldt, J.; Legge, R.M.; Ma, F.; Hua, K.; Sinha, R.; Peterson, D.A.; Walter, J.; Benson, A.K.; et al. Host genetics and diet, but not immunoglobulin A expression, converge to shape compositional features of the gut microbiome in an advanced intercross population of mice. Genome Biol. 2014, 15, 552. [Google Scholar] [CrossRef] [Green Version]
- Elhag, O.; Zhou, D.; Song, Q.; Soomro, A.A.; Cai, M.; Zheng, L.; Yu, Z.; Zhang, J. Screening, expression, purification and functional characterization of novel antimicrobial peptide genes from Hermetia illucens (L.). PLoS ONE 2017, 12, e0169582. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Yoe, S.M. A novel cecropin-like peptide from black soldier fly, Hermetia illucens: Isolation, structural and functional characterization. Entomol. Res. 2017, 47, 115–124. [Google Scholar] [CrossRef]
- Park, S.; Yoe, S.M. Defensin-like peptide3 from black solder fly: Identification, characterization, and key amino acids for anti-Gram-negative bacteria. Entomol. Res. 2017, 47, 41–47. [Google Scholar] [CrossRef]
- Vogel, H.; Müller, A.; Heckel, D.G.; Gutzeit, H.; Vilcinskas, A. Nutritional immunology: Diversification and diet-dependent expression of antimicrobial peptides in the black soldier fly Hermetia illucens. Dev. Comp. Immunol. 2018, 78, 141–148. [Google Scholar] [CrossRef]
- Shin, H.S.; Park, S. Novel attacin from Hermetia illucens: cDNA cloning, characterization, and antibacterial properties. Prep. Biochem. Biotechnol. 2019, 49, 279–285. [Google Scholar] [CrossRef]
- Shafquat, A.; Joice, R.; Simmons, S.L.; Huttenhower, C. Functional and phylogenetic assembly of microbial communities in the human microbiome. Trends Microbiol. 2014, 22, 261–266. [Google Scholar] [CrossRef] [Green Version]
- Stothart, M.R.; Greuel, R.J.; Gavriliuc, S.; Henry, A.; Wilson, A.J.; McLoughlin, P.D.; Poissant, J. Bacterial dispersal and drift drive microbiome diversity patterns within a population of feral hindgut fermenters. Mol. Ecol. 2021, 30, 555–571. [Google Scholar] [CrossRef] [PubMed]
- Nemergut, D.R.; Schmidt, S.K.; Fukami, T.; O’Neill, S.P.; Bilinski, T.M.; Stanish, L.F.; Knelman, J.E.; Darcy, J.L.; Lynch, R.C.; Wickey, P.; et al. Patterns and processes of microbial community assembly. Microbiol. Mol. Biol. Rev. 2013, 77, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.-X.; Song, Z.-R.; Huo, S.-M.; Yang, K.; Hong, X.-Y. Variation in the microbiome of the spider mite Tetranychus truncatus with sex, instar and endosymbiont infection. FEMS Microbiol. Ecol. 2020, 96, fiaa004. [Google Scholar] [CrossRef]
- Minard, G.; Mavingui, P.; Moro, C. Diversity and function of bacterial microbiota in the mosquito holobiont. Parasit. Vectors 2013, 6, 146. [Google Scholar] [CrossRef] [Green Version]
- Ross, E.M.; Moate, P.J.; Marett, L.C.; Cocks, B.G.; Hayes, B.J. Metagenomic predictions: From microbiome to complex health and environmental phenotypes in humans and cattle. PLoS ONE 2013, 8, e73056. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Bonder, M.J.; Cenit, M.C.; Tigchelaar, E.F.; Maatman, A.; Dekens, J.A.M.; Brandsma, E.; Marczynska, J.; Imhann, F.; Weersma, R.K.; et al. The gut microbiome contributes to a substantial proportion of the variation in blood lipids. Circ. Res. 2015, 117, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Khanal, P.; Maltecca, C.; Schwab, C.; Fix, J.; Bergamaschi, M.; Tiezzi, F. Modeling host-microbiome interactions for the prediction of meat quality and carcass composition traits in swine. Genet. Sel. Evol. 2020, 52, 41. [Google Scholar] [CrossRef] [PubMed]
- Estellé, J. Benefits from the joint analysis of host genomes and metagenomes: Select the holobiont. J. Anim. Breed. Genet. 2019, 136, 75–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruokolainen, L.; Ikonen, S.; Makkonen, H.; Hanski, I. Larval growth rate is associated with the composition of the gut microbiota in the Glanville fritillary butterfly. Oecologia 2016, 181, 895–903. [Google Scholar] [CrossRef]
- Chaston, J.M.; Dobson, A.J.; Newell, P.D.; Douglas, A.E. Host genetic control of the microbiota mediates the Drosophila nutritional phenotype. Appl. Environ. Microbiol. 2016, 82, 671–679. [Google Scholar] [CrossRef] [Green Version]
- Storelli, G.; Defaye, A.; Erkosar, B.; Hols, P.; Royet, J.; Leulier, F. Lactobacillus plantarum promotes Drosophila systemic growth by modulating hormonal signals through TOR-dependent nutrient sensing. Cell Metab. 2011, 14, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Macfarlane, G.T.; Allison, C. Utilisation of protein by human gut bacteria. FEMS Microbiol. Lett. 1986, 2, 19–24. [Google Scholar] [CrossRef]
- Salwan, R.; Sharma, V. The role of actinobacteria in the production of industrial enzymes. In New and Future Developments in Microbial Biotechnology and Bioengineering; Elsevier: Palampur, India, 2018; pp. 165–177. [Google Scholar]
- Vera-Ponce de León, A.; Jahnes, B.C.; Duan, J.; Camuy-Vélez, L.A.; Sabree, Z.L. Cultivable, host-specific Bacteroidetes symbionts exhibit diverse polysaccharolytic strategies. Appl. Environ. Microbiol. 2020, 86, e00091-20. [Google Scholar] [CrossRef] [PubMed]
- Kaltenpoth, M. Actinobacteria as mutualists: General healthcare for insects? Trends Microbiol. 2009, 17, 529–535. [Google Scholar] [CrossRef]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Shin, N.; Whon, T.W.; Bae, J. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Pindling, S.; Azulai, D.; Zheng, B.; Dahan, D.; Perron, G.G. Dysbiosis and early mortality in zebrafish larvae exposed to subclinical concentrations of streptomycin. FEMS Microbiol. Lett. 2018, 365, fny188. [Google Scholar] [CrossRef]
- Flury, P.; Aellen, N.; Ruffner, B.; Péchy-Tarr, M.; Fataar, S.; Metla, Z.; Dominguez-Ferreras, A.; Bloemberg, G.; Frey, J.; Goesmann, A.; et al. Insect pathogenicity in plant-beneficial pseudomonads: Phylogenetic distribution and comparative genomics. ISME J. 2016, 10, 2527–2542. [Google Scholar] [CrossRef] [PubMed]
- Vilanova, C.; Baixeras, J.; Latorre, A.; Porcar, M. The generalist inside the specialist: Gut bacterial communities of two insect species feeding on toxic plants are dominated by Enterococcus sp. Front. Microbiol. 2016, 7, 1005. [Google Scholar] [CrossRef]
- Gayatri Priya, N.; Ojha, A.; Kajla, M.K.; Raj, A.; Rajagopal, R. Host plant induced variation in gut bacteria of Helicoverpa armigera. PLoS ONE 2012, 7, e30768. [Google Scholar] [CrossRef] [Green Version]
- Tarkhani, R.; Imani, A.; Hoseinifar, S.H.; Sarvi Moghanlou, K.; Manaffar, R. The effects of host-associated Enterococcus faecium CGMCC1.2136 on serum immune parameters, digestive enzymes activity and growth performance of the Caspian roach (Rutilus rutilus caspicus) fingerlings. Aquaculture 2020, 519, 734741. [Google Scholar] [CrossRef]
- Dubin, K.; Pamer, E.G. Enterococci and their interactions with the intestinal microbiome. Microbiol. Spectr. 2017, 5, 309–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Teh, B.S.; Sun, C.; Hu, S.; Lu, X.; Boland, W.; Shao, Y. Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis. Sci. Rep. 2016, 6, 29505. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, C.; Milanović, V.; Cardinali, F.; Aquilanti, L.; Clementi, F.; Osimani, A. Current knowledge on the microbiota of edible insects intended for human consumption: A state-of-the-art review. Food Res. Int. 2019, 125, 108527. [Google Scholar] [CrossRef] [PubMed]
- Manos, J.; Belas, R. The genera Proteus, Providencia, and Morganella. In The Prokaryotes; Springer: New York, NY, USA, 2006; pp. 245–269. [Google Scholar]
- Salas, B.; Conway, H.E.; Schuenzel, E.L.; Hopperstad, K.; Vitek, C.; Vacek, D.C. Morganella morganii (Enterobacteriales: Enterobacteriaceae) is a lethal pathogen of Mexican fruit fly (Diptera: Tephritidae) larvae. Florida Entomol. 2017, 100, 743–751. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Partition | Df | SS | R2 | Pseudo-F | p |
|---|---|---|---|---|---|
| Background (G) | 1 | 0.357 | 0.078 | 3.053 | 0.009 |
| Feed (E) | 3 | 1.714 | 0.374 | 4.885 | <0.001 |
| Background × Feed (G × E) | 3 | 0.637 | 0.139 | 1.815 | 0.031 |
| Residual | 16 | 1.872 | 0.409 | ||
| Total | 23 | 4.580 | 1.000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greenwood, M.P.; Hull, K.L.; Brink-Hull, M.; Lloyd, M.; Rhode, C. Feed and Host Genetics Drive Microbiome Diversity with Resultant Consequences for Production Traits in Mass-Reared Black Soldier Fly (Hermetia illucens) Larvae. Insects 2021, 12, 1082. https://doi.org/10.3390/insects12121082
Greenwood MP, Hull KL, Brink-Hull M, Lloyd M, Rhode C. Feed and Host Genetics Drive Microbiome Diversity with Resultant Consequences for Production Traits in Mass-Reared Black Soldier Fly (Hermetia illucens) Larvae. Insects. 2021; 12(12):1082. https://doi.org/10.3390/insects12121082
Chicago/Turabian StyleGreenwood, Matthew P., Kelvin L. Hull, Marissa Brink-Hull, Melissa Lloyd, and Clint Rhode. 2021. "Feed and Host Genetics Drive Microbiome Diversity with Resultant Consequences for Production Traits in Mass-Reared Black Soldier Fly (Hermetia illucens) Larvae" Insects 12, no. 12: 1082. https://doi.org/10.3390/insects12121082