Pharmacogenomic Impact of CYP2C19 Variation on Clopidogrel Therapy in Precision Cardiovascular Medicine


Abstract
:1. Introduction
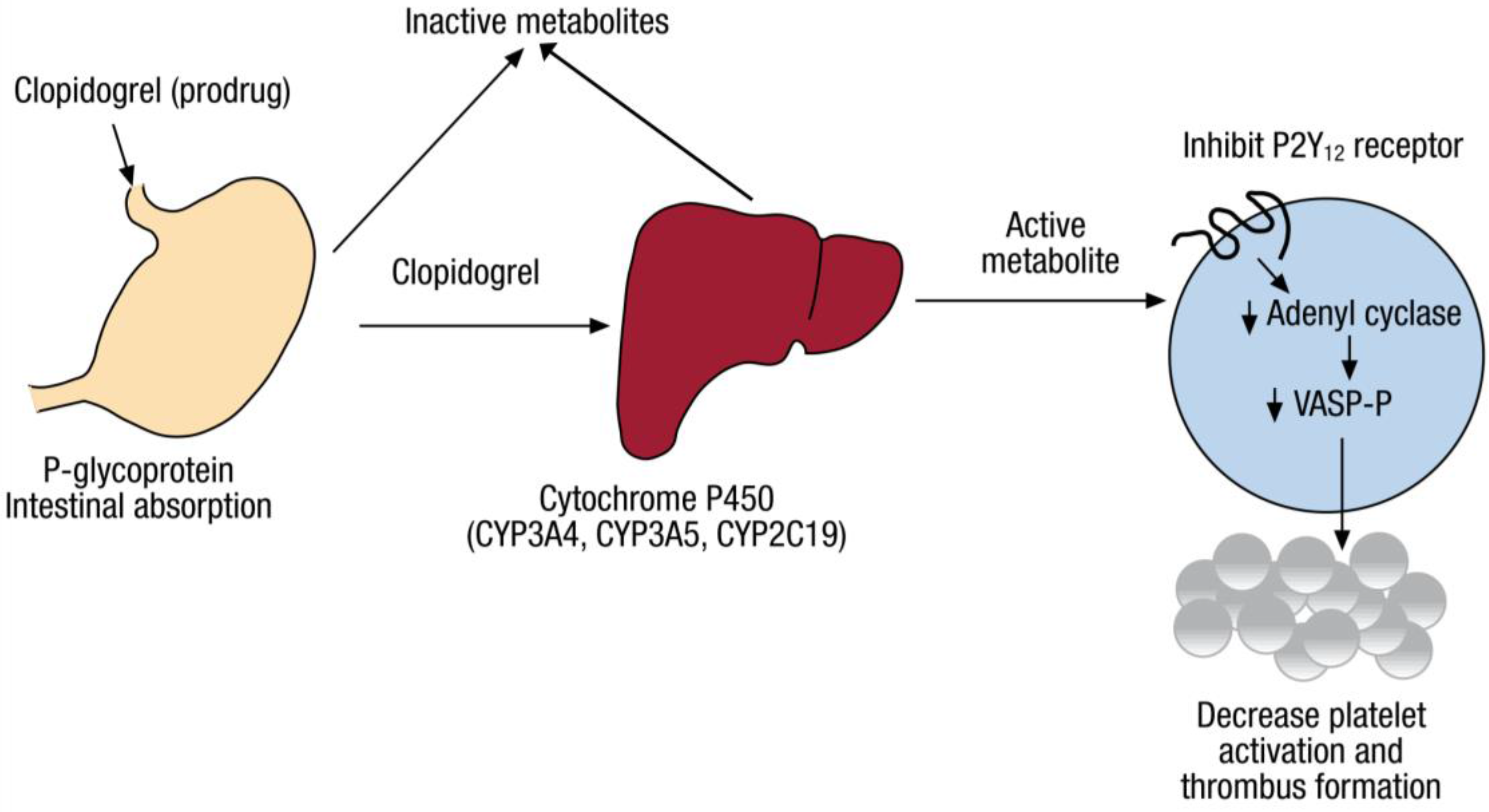
2. Pharmacogenomics of Clopidogrel Response Variation
2.1. CYP2C19 Variation
2.1.1. Loss-of-Function CYP2C19 Variants
2.1.2. Gain-of-Function CYP2C19 Variant
2.1.3. Linkage Disequilibrium
2.1.4. Multiple Variants
3. Pharmacogenomics Era
3.1. Pharmacokinetic Phenotypes
3.2. Impact on Platelet Function
3.3. Impact on Clopidogrel Metabolite Plasma Concentration
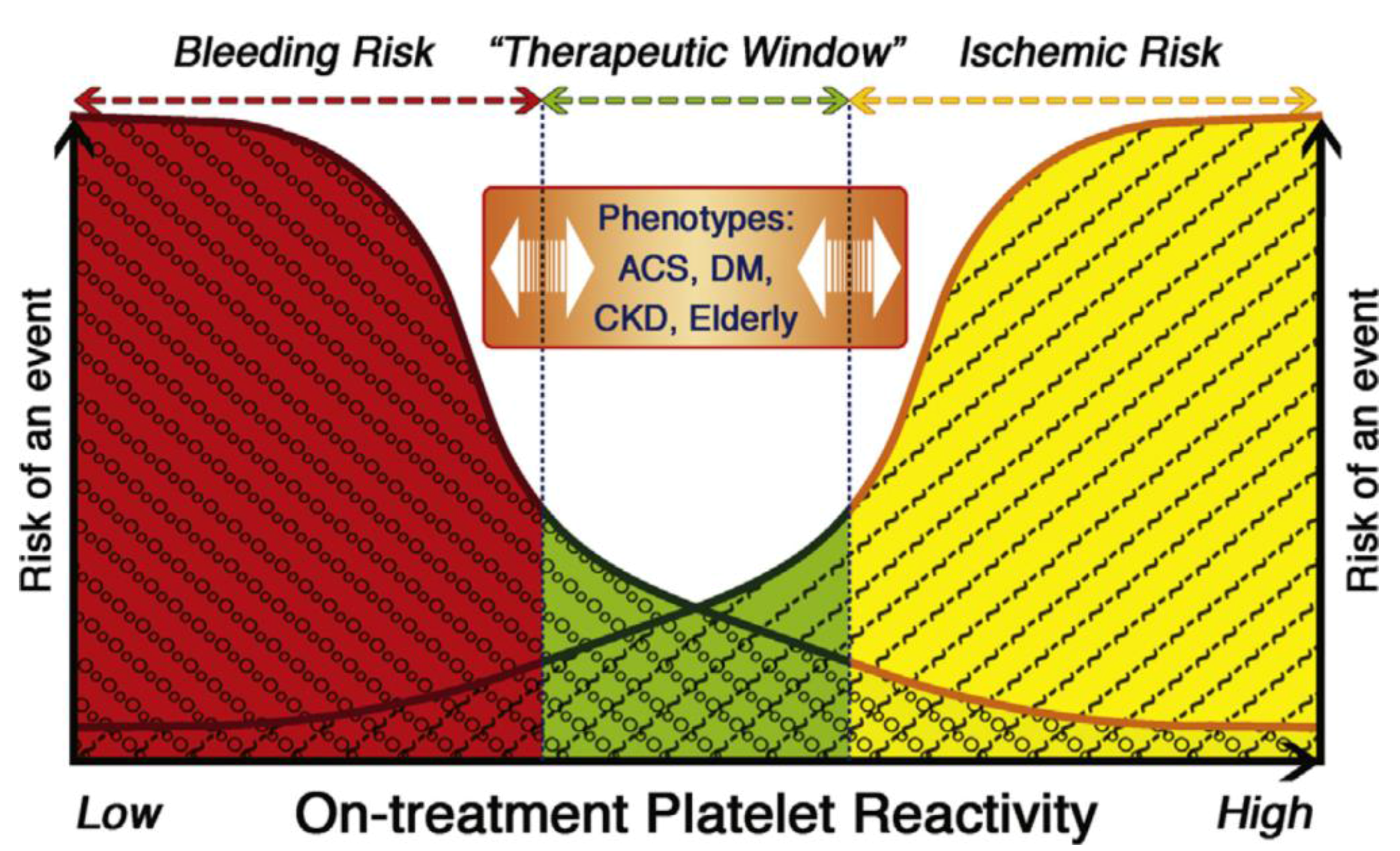
3.4. Impact on Outcomes/Events
4. Regulators of Response
4.1. Variation by Ethnicity
4.2. Drug-Drug Interactions
4.3. Conventional Regulators of Response
4.3.1. Adherence
4.3.2. Ischemic Heart Disease Risk Factors
Sociodemographic Characteristics
Lifestyle Habits
Comorbid Conditions
5. Multi-Omic Precision Medicine Approach to Response Regulation
5.1. Transcriptomics
5.2. Epigenomics and Exposomics
5.3. miRNA Regulomics
5.4. Proteomics
5.5. Metabolomics and Microbiomics
5.6. Mathematical, Computational, and Molecular Modeling
6. Toward Implementation in Clinical Practice
Funding
Author Contributions
Conflicts of Interest
References
- Fox, C.S.; Hall, J.L.; Arnett, D.K.; Ashley, E.A.; Delles, C.; Engler, M.B.; Freeman, M.W.; Johnson, J.A.; Lanfear, D.E.; Liggett, S.B.; et al. Future translational applications from the contemporary genomics era: A scientific statement from the American Heart Association. Circulation 2015, 131, 1715–1736. [Google Scholar] [CrossRef] [PubMed]
- O’Gara, P.T.; Kushner, F.G.; Ascheim, D.D.; Casey, D.E.; Chung, M.K.; de Lemos, J.A.; Ettinger, S.M.; Fang, J.C.; Fesmire, F.M.; Franklin, B.A.; et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013, 127, e362–e425. [Google Scholar] [CrossRef] [PubMed]
- Levine, G.N.; Bates, E.R.; Blankenship, J.C.; Bailey, S.R.; Bittl, J.A.; Cercek, B.; Chambers, C.E.; Ellis, S.G.; Guyton, R.A.; Hollenberg, S.M.; et al. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. Circulation 2011, 124, 2574–2609. [Google Scholar] [CrossRef] [PubMed]
- Rafique, A.M.; Nayyar, P.; Wang, T.Y.; Mehran, R.; Baber, U.; Berger, P.B.; Tobis, J.; Currier, J.; Dave, R.H.; Henry, T.D. Optimal P2Y12 Inhibitor in Patients With ST-Segment Elevation Myocardial Infarction Undergoing Primary Percutaneous Coronary Intervention: A Network Meta-Analysis. JACC Cardiovasc. Interv. 2016, 9, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.S.; Bhatt, D.L.; Cannon, C.P.; Chen, Z.; Jiang, L.; Jones, J.B.; Mehta, S.R.; Sabatine, M.S.; Steinhubl, S.R.; Topol, E.J.; et al. The relative efficacy and safety of clopidogrel in women and men a sex-specific collaborative meta-analysis. J. Am. Coll. Cardiol. 2009, 54, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Torpy, J.M.; Lynm, C.; Glass, R.M. JAMA patient page. Percutaneous coronary intervention. JAMA 2004, 291, 778. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.G.; Gruntowicz, D.; Chua, T.; Morlock, R.J. Financial analysis of CYP2C19 genotyping in patients receiving dual antiplatelet therapy following acute coronary syndrome and percutaneous coronary intervention. J. Manag. Care Spec. Pharm. 2015, 21, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.P.; Shuldiner, A.R. Clopidogrel pharmacogenetics: Beyond candidate genes and genome-wide association studies. Clin. Pharmacol. Ther. 2017, 101, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Oprea, A.D.; Popescu, W.M. P2Y12 receptor inhibitors in acute coronary syndromes: What is new on the horizon? Cardiol. Res. Pract. 2013, 2013, 195456. [Google Scholar] [CrossRef] [PubMed]
- Taubert, D.; Kastrati, A.; Harlfinger, S.; Gorchakova, O.; Lazar, A.; von Beckerath, N.; Schömig, A.; Schömig, E. Pharmacokinetics of clopidogrel after administration of a high loading dose. Thromb. Haemost. 2004, 92, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.M.; Sheau Chin, L.; Azri Mohamed Noor, D.; Sk Abdul Kader, M.A.; Kah Hay, Y.; Ibrahim, B. The Personalization of Clopidogrel Antiplatelet Therapy: The role of integrative pharmacogenetics and pharmacometabolomics. Cardiol. Res. Pract. 2017, 2017, 8062796. [Google Scholar] [CrossRef] [PubMed]
- Schrör, K. Clinical pharmacology of the adenosine diphosphate (ADP) receptor antagonist, clopidogrel. Vasc. Med. 1998, 3, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Shepard, R.W. Pharmacology: Antiplatelet and antithrombin therapy in acute coronary syndromes. J. Cardiovasc. Nurs. 2000, 15, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Shameem, R.; Hamid, M.; Randhawa, A.; Spaccavento, C.; Garatt, K. P2Y12 Antagonists: Pharmacology, efficacy and patient considerations. J. Cardiovasc. Dis. 2014, 2, 91–100. [Google Scholar]
- Pereira, N.L.; Geske, J.B.; Mayr, M.; Shah, S.H.; Rihal, C.S. Pharmacogenetics of clopidogrel: An unresolved issue. Circ. Cardiovasc. Genet. 2016, 9, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.; Bhatt, D.L.; Bergougnan, L.; Farenc, C.; Pearson, K.; Perrin, L.; Vicaut, E.; Lacreta, F.; Hurbin, F.; Dubar, M. Genetic polymorphisms and the impact of a higher clopidogrel dose regimen on active metabolite exposure and antiplatelet response in healthy subjects. Clin. Pharmacol. Ther. 2011, 90, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Meadows, T.A.; Bhatt, D.L. Clinical aspects of platelet inhibitors and thrombus formation. Circ. Res. 2007, 100, 1261–1275. [Google Scholar] [CrossRef] [PubMed]
- Davì, G.; Patrono, C. Platelet activation and atherothrombosis. N. Engl. J. Med. 2007, 357, 2482–2494. [Google Scholar] [CrossRef] [PubMed]
- Falk, E. Coronary thrombosis: Pathogenesis and clinical manifestations. Am. J. Cardiol. 1991, 68, 28B–35B. [Google Scholar] [CrossRef]
- Fuster, V.; Badimon, L.; Badimon, J.J.; Chesebro, J.H. The pathogenesis of coronary artery disease and the acute coronary syndromes (2). N. Engl. J. Med. 1992, 326, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, Z.M. Platelets in atherothrombosis. Nat. Med. 2002, 8, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.E.; Iafrati, H.F. Use of genetics and transcriptomics in the diagnosis and treatment of coronary artery disease. Rev. Esp. Cardiol. 2010, 63, 1123–1126. [Google Scholar] [CrossRef]
- Hulot, J.S.; Collet, J.P.; Silvain, J.; Pena, A.; Bellemain-Appaix, A.; Barthélémy, O.; Cayla, G.; Beygui, F.; Montalescot, G. Cardiovascular risk in clopidogrel-treated patients according to cytochrome P450 2C19*2 loss-of-function allele or proton pump inhibitor coadministration: A systematic meta-analysis. J. Am. Coll. Cardiol. 2010, 56, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Gurbel, P.A.; Bliden, K.P.; Hiatt, B.L.; O’Connor, C.M. Clopidogrel for coronary stenting: Response variability, drug resistance, and the effect of pretreatment platelet reactivity. Circulation 2003, 107, 2908–2913. [Google Scholar] [CrossRef] [PubMed]
- Gross, L.; Sibbing, D. Should We Perform Genetic Testing on Antiplatelet Therapy? 2017. Available online: https://www.acc.org/latest-in-cardiology/articles/2017/07/10/09/17/should-we-perform-genetic-testing-on-antiplatelet-therapy?w_nav=CI (accessed on 20 September 2017).
- Shuldiner, A.R.; O’Connell, J.R.; Bliden, K.P.; Gandhi, A.; Ryan, K.; Horenstein, R.B.; Damcott, C.M.; Pakyz, R.; Tantry, U.S.; Gibson, Q.; et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009, 302, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Cappola, T.P.; Margulies, K.B. Functional genomics applied to cardiovascular medicine. Circulation 2011, 124, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. The Metabolomic Paradigm of Pharmacogenomics in Complex Disorders. Metabolomics 2012, 2, e119. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: The past, present and future. Trends Pharmacol. Sci. 2004, 25, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M.; Sim, S.C.; Gomez, A.; Rodriguez-Antona, C. Influence of cytochrome P450 polymorphisms on drug therapies: Pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol. Ther. 2007, 116, 496–526. [Google Scholar] [CrossRef] [PubMed]
- Speed, W.C.; Kang, S.P.; Tuck, D.P.; Harris, L.N.; Kidd, K.K. Global variation in CYP2C8-CYP2C9 functional haplotypes. Pharmacogenom. J. 2009, 9, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.Q.; Wang, D.G.; Yang, H.; Cao, H. Cytochrome P450 CYP 2C19*2 associated with adverse 1-year cardiovascular events in patients with acute coronary syndrome. PLoS ONE 2015, 10, e0132561. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.W.; Koo, B.K.; Zhang, S.Y.; Park, K.W.; Cho, J.Y.; Jang, I.J.; Lee, D.S.; Sohn, D.W.; Lee, M.M.; Kim, H.S. Increased risk of atherothrombotic events associated with cytochrome P450 3A5 polymorphism in patients taking clopidogrel. CMAJ 2006, 174, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.T.; Close, S.L.; Iturria, S.J.; Payne, C.D.; Farid, N.A.; Ernest, C.S.; Lachno, D.R.; Salazar, D.; Winters, K.J. Common polymorphisms of CYP2C19 and CYP2C9 affect the pharmacokinetic and pharmacodynamic response to clopidogrel but not prasugrel. J. Thromb. Haemost. 2007, 5, 2429–2436. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Xu, J.; Li, X.; Zhang, H.; Hu, J.; Fang, R.; Chen, X. ABCB1 C3435T polymorphism and response to clopidogrel treatment in coronary artery disease (CAD) patients: A meta-analysis. PLoS ONE 2012, 7, e46366. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.P.; Horenstein, R.B.; Ryan, K.; O’Connell, J.R.; Gibson, Q.; Mitchell, B.D.; Tanner, K.; Chai, S.; Bliden, K.P.; Tantry, U.S.; et al. The functional G143E variant of carboxylesterase 1 is associated with increased clopidogrel active metabolite levels and greater clopidogrel response. Pharmacogenet. Genom. 2013, 23, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Staritz, P.; Kurz, K.; Stoll, M.; Giannitsis, E.; Katus, H.A.; Ivandic, B.T. Platelet reactivity and clopidogrel resistance are associated with the H2 haplotype of the P2Y12-ADP receptor gene. Int. J. Cardiol. 2009, 133, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Bains, R.K. African variation at Cytochrome P450 genes: Evolutionary aspects and the implications for the treatment of infectious diseases. Evol. Med. Public Health 2013, 2013, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.S.; Kim, D. Polymorphic metabolism by functional alterations of human cytochrome P450 enzymes. Arch. Pharm. Res. 2011, 34, 1799–1816. [Google Scholar] [CrossRef] [PubMed]
- Kazui, M.; Nishiya, Y.; Ishizuka, T.; Hagihara, K.; Farid, N.A.; Okazaki, O.; Ikeda, T.; Kurihara, A. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab. Dispos. 2010, 38, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D.R.; Dehmer, G.J.; Kaul, S.; Leifer, D.; O’Gara, P.T.; Stein, C.M. ACCF/AHA clopidogrel clinical alert: Approaches to the FDA “boxed warning”: A report of the American College of Cardiology Foundation Task Force on clinical expert consensus documents and the American Heart Association endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. J. Am. Coll. Cardiol. 2010, 56, 321–341. [Google Scholar] [CrossRef] [PubMed]
- Maseneni, S.; Donzelli, M.; Taegtmeyer, A.B.; Brecht, K.; Krähenbühl, S. Toxicity of clopidogrel and ticlopidine on human myeloid progenitor cells: Importance of metabolites. Toxicology 2012, 299, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Trenk, D.; Hochholzer, W.; Fromm, M.F.; Chialda, L.E.; Pahl, A.; Valina, C.M.; Stratz, C.; Schmiebusch, P.; Bestehorn, H.P.; Büttner, H.J.; et al. Cytochrome P450 2C19 681G>A polymorphism and high on-clopidogrel platelet reactivity associated with adverse 1-year clinical outcome of elective percutaneous coronary intervention with drug-eluting or bare-metal stents. J. Am. Coll. Cardiol. 2008, 51, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Mega, J.L.; Close, S.L.; Wiviott, S.D.; Shen, L.; Hockett, R.D.; Brandt, J.T.; Walker, J.R.; Antman, E.M.; Macias, W.; Braunwald, E.; et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N. Engl. J. Med. 2009, 360, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.J.; Fernandez-Ortiz, A.; Bernardo, E.; Alfonso, F.; Macaya, C.; Bass, T.A.; Costa, M.A. Variability in individual responsiveness to clopidogrel: Clinical implications, management, and future perspectives. J. Am. Coll. Cardiol. 2007, 49, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Sanford, J.C.; Guo, Y.; Sadee, W.; Wang, D. Regulatory polymorphisms in CYP2C19 affecting hepatic expression. Drug Metabol. Drug Interact. 2013, 28, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Helsby, N.A.; Burns, K.E. Molecular mechanisms of genetic variation and transcriptional regulation of CYP2C19. Front. Genet. 2012, 3, 206. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M. CYP2C19 Allele Nomenclature. Available online: http://www.cypalleles.ki.se/cyp2c19.htm (accessed on 20 September 2017).
- Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Worldwide Distribution of Cytochrome P450 Alleles: A Meta-analysis of Population-scale Sequencing Projects. Clin. Pharmacol. Ther. 2017, 102, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Romkes, M.; Faletto, M.B.; Blaisdell, J.A.; Raucy, J.L.; Goldstein, J.A. Cloning and expression of complementary DNAs for multiple members of the human cytochrome P450IIC subfamily. Biochemistry 1991, 30, 3247–3255. [Google Scholar] [CrossRef] [PubMed]
- De Morais, S.M.; Wilkinson, G.R.; Blaisdell, J.; Nakamura, K.; Meyer, U.A.; Goldstein, J.A. The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J. Biol. Chem. 1994, 269, 15419–15422. [Google Scholar] [PubMed]
- Ibeanu, G.C.; Goldstein, J.A.; Meyer, U.; Benhamou, S.; Bouchardy, C.; Dayer, P.; Ghanayem, B.I.; Blaisdell, J. Identification of new human CYP2C19 alleles (CYP2C19*6 and CYP2C19*2B) in a Caucasian poor metabolizer of mephenytoin. J. Pharmacol. Exp. Ther. 1998, 286, 1490–1495. [Google Scholar] [PubMed]
- Fukushima-Uesaka, H.; Saito, Y.; Maekawa, K.; Ozawa, S.; Hasegawa, R.; Kajio, H.; Kuzuya, N.; Yasuda, K.; Kawamoto, M.; Kamatani, N.; et al. Genetic variations and haplotypes of CYP2C19 in a Japanese population. Drug Metab. Pharmacokinet. 2005, 20, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, W.Y.; Kim, H.; Shon, J.H.; Lee, S.S.; Shin, J.G. Identification of new CYP2C19 variants exhibiting decreased enzyme activity in the metabolism of S-mephenytoin and omeprazole. Drug Metab. Dispos. 2009, 37, 2262–2269. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, C.R.; Devendran, A.; Jayaraman, M.; Mannu, J.; Mathur, P.P.; Gopal, S.D.; Rajagopal, K.; Chandrasekaran, A. Influence of the genetic polymorphisms in the 5′ flanking and exonic regions of CYP2C19 on proguanil oxidation. Drug Metab. Pharmacokinet. 2009, 24, 537–548. [Google Scholar] [CrossRef] [PubMed]
- De Morais, S.M.; Wilkinson, G.R.; Blaisdell, J.; Meyer, U.A.; Nakamura, K.; Goldstein, J.A. Identification of a new genetic defect responsible for the polymorphism of (S)-mephenytoin metabolism in Japanese. Mol. Pharmacol. 1994, 46, 594–598. [Google Scholar] [PubMed]
- Ferguson, R.J.; De Morais, S.M.; Benhamou, S.; Bouchardy, C.; Blaisdell, J.; Ibeanu, G.; Wilkinson, G.R.; Sarich, T.C.; Wright, J.M.; Dayer, P.; et al. A new genetic defect in human CYP2C19: Mutation of the initiation codon is responsible for poor metabolism of S-mephenytoin. J. Pharmacol. Exp. Ther. 1998, 284, 356–361. [Google Scholar] [PubMed]
- Scott, S.A.; Martis, S.; Peter, I.; Kasai, Y.; Kornreich, R.; Desnick, R.J. Identification of CYP2C19*4B: Pharmacogenetic implications for drug metabolism including clopidogrel responsiveness. Pharmacogenom. J. 2012, 12, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.S.; Goldstein, J.A.; Xie, H.G.; Blaisdell, J.; Wang, W.; Jiang, C.H.; Yan, F.X.; He, N.; Huang, S.L.; Xu, Z.H.; et al. Differences in the incidence of the CYP2C19 polymorphism affecting the S-mephenytoin phenotype in Chinese Han and Bai populations and identification of a new rare CYP2C19 mutant allele. J. Pharmacol. Exp. Ther. 1997, 281, 604–609. [Google Scholar] [PubMed]
- Ibeanu, G.C.; Blaisdell, J.; Ghanayem, B.I.; Beyeler, C.; Benhamou, S.; Bouchardy, C.; Wilkinson, G.R.; Dayer, P.; Daly, A.K.; Goldstein, J.A. An additional defective allele, CYP2C19*5, contributes to the S-mephenytoin poor metabolizer phenotype in Caucasians. Pharmacogenetics 1998, 8, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Ibeanu, G.C.; Blaisdell, J.; Ferguson, R.J.; Ghanayem, B.I.; Brosen, K.; Benhamou, S.; Bouchardy, C.; Wilkinson, G.R.; Dayer, P.; Goldstein, J.A. A novel transversion in the intron 5 donor splice junction of CYP2C19 and a sequence polymorphism in exon 3 contribute to the poor metabolizer phenotype for the anticonvulsant drug S-mephenytoin. J. Pharmacol. Exp. Ther. 1999, 290, 635–640. [Google Scholar] [PubMed]
- Blaisdell, J.; Mohrenweiser, H.; Jackson, J.; Ferguson, S.; Coulter, S.; Chanas, B.; Xi, T.; Ghanayem, B.; Goldstein, J.A. Identification and functional characterization of new potentially defective alleles of human CYP2C19. Pharmacogenetics 2002, 12, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Morita, J.; Kobayashi, K.; Wanibuchi, A.; Kimura, M.; Irie, S.; Ishizaki, T.; Chiba, K. A novel single nucleotide polymorphism (SNP) of the CYP2C19 gene in a Japanese subject with lowered capacity of mephobarbital 4′-hydroxylation. Drug Metab. Pharmacokinet. 2004, 19, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Sim, S.C.; Risinger, C.; Dahl, M.L.; Aklillu, E.; Christensen, M.; Bertilsson, L.; Ingelman-Sundberg, M. A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin. Pharmacol. Ther. 2006, 79, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Matimba, A.; Del-Favero, J.; Van Broeckhoven, C.; Masimirembwa, C. Novel variants of major drug-metabolising enzyme genes in diverse African populations and their predicted functional effects. Hum. Genom. 2009, 3, 169–190. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Yu, X.M.; Lin, H.B.; Wang, L.; Yun, Q.Z.; Hu, S.N.; Wang, D.M. Genetic polymorphism, linkage disequilibrium, haplotype structure and novel allele analysis of CYP2C19 and CYP2D6 in Han Chinese. Pharmacogenom. J. 2009, 9, 380–394. [Google Scholar] [CrossRef] [PubMed]
- Drögemöller, B.I.; Wright, G.E.; Niehaus, D.J.; Koen, L.; Malan, S.; Da Silva, D.M.; Hillermann-Rebello, R.; La Grange, A.M.; Venter, M.; Warnich, L. Characterization of the genetic profile of CYP2C19 in two South African populations. Pharmacogenomics 2010, 11, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.A.; Sangkuhl, K.; Stein, C.M.; Hulot, J.S.; Mega, J.L.; Roden, D.M.; Klein, T.E.; Sabatine, M.S.; Johnson, J.A.; Shuldiner, A.R.; et al. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin. Pharmacol. Ther. 2013, 94, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Luzum, J.A.; Pakyz, R.E.; Elsey, A.R.; Haidar, C.E.; Peterson, J.F.; Whirl-Carrillo, M.; Handelman, S.K.; Palmer, K.; Pulley, J.M.; Beller, M.; et al. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: Outcomes and Metrics of Pharmacogenetic Implementations across Diverse Healthcare Systems. Clin. Pharmacol. Ther. 2017, 102, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Sistonen, J.; Fuselli, S.; Palo, J.U.; Chauhan, N.; Padh, H.; Sajantila, A. Pharmacogenetic variation at CYP2C9, CYP2C19, and CYP2D6 at global and microgeographic scales. Pharmacogenet. Genom. 2009, 19, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Uppugunduri, C.R.; Daali, Y.; Desmeules, J.; Dayer, P.; Krajinovic, M.; Ansari, M. Transcriptional regulation of CYP2C19 and its role in altered enzyme activity. Curr. Drug Metab. 2012, 13, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.A.; Sangkuhl, K.; Gardner, E.E.; Stein, C.M.; Hulot, J.S.; Johnson, J.A.; Roden, D.M.; Klein, T.E.; Shuldiner, A.R.; Consortium, C.P.I. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450-2C19 (CYP2C19) genotype and clopidogrel therapy. Clin. Pharmacol. Ther. 2011, 90, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Sibbing, D.; Koch, W.; Gebhard, D.; Schuster, T.; Braun, S.; Stegherr, J.; Morath, T.; Schömig, A.; von Beckerath, N.; Kastrati, A. Cytochrome 2C19*17 allelic variant, platelet aggregation, bleeding events, and stent thrombosis in clopidogrel-treated patients with coronary stent placement. Circulation 2010, 121, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Wallentin, L.; James, S.; Storey, R.F.; Armstrong, M.; Barratt, B.J.; Horrow, J.; Husted, S.; Katus, H.; Steg, P.G.; Shah, S.H.; et al. Effect of CYP2C19 and ABCB1 single nucleotide polymorphisms on outcomes of treatment with ticagrelor versus clopidogrel for acute coronary syndromes: A genetic substudy of the PLATO trial. Lancet 2010, 376, 1320–1328. [Google Scholar] [CrossRef]
- Simon, T.; Verstuyft, C.; Mary-Krause, M.; Quteineh, L.; Drouet, E.; Méneveau, N.; Steg, P.G.; Ferrières, J.; Danchin, N.; Becquemont, L.; et al. Genetic determinants of response to clopidogrel and cardiovascular events. N. Engl. J. Med. 2009, 360, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Gurbel, P.A.; Shuldiner, A.R.; Bliden, K.P.; Ryan, K.; Pakyz, R.E.; Tantry, U.S. The relation between CYP2C19 genotype and phenotype in stented patients on maintenance dual antiplatelet therapy. Am. Heart J. 2011, 161, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Gladding, P.; Webster, M.; Zeng, I.; Farrell, H.; Stewart, J.; Ruygrok, P.; Ormiston, J.; El-Jack, S.; Armstrong, G.; Kay, P.; et al. The pharmacogenetics and pharmacodynamics of clopidogrel response: An analysis from the PRINC (Plavix Response in Coronary Intervention) trial. JACC Cardiovasc. Interv. 2008, 1, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.M.; Ohlsson, S.; Pedersen, R.S.; Mwinyi, J.; Ingelman-Sundberg, M.; Eliasson, E.; Bertilsson, L. Increased omeprazole metabolism in carriers of the CYP2C19*17 allele; a pharmacokinetic study in healthy volunteers. Br. J. Clin. Pharmacol. 2008, 65, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Stephens, S.; Horenstein, R.; O’Connell, J.; Ryan, K.; Peer, C.; Figg, W.; Spencer, S.; Pacanowski, M.; Mitchell, B.; et al. The CYP2C19*17 variant is not independently associated with clopidogrel response. J. Thromb. Haemost. 2013, 11, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Skierka, J.M.; Black, J.L. Analysis of compound heterozygous CYP2C19 genotypes to determine cis and trans configurations. Pharmacogenomics 2014, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.S.; Prasad, B.; Shirasaka, Y.; Fohner, A.; Finkelstein, D.; Fan, Y.; Wang, S.; Wu, G.; Aklillu, E.; Sim, S.C.; et al. The CYP2C19 Intron 2 Branch Point SNP is the Ancestral Polymorphism Contributing to the Poor Metabolizer Phenotype in Livers with CYP2C19*35 and CYP2C19*2 Alleles. Drug Metab. Dispos. 2015, 43, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Mega, J.L.; Close, S.L.; Wiviott, S.D.; Shen, L.; Walker, J.R.; Simon, T.; Antman, E.M.; Braunwald, E.; Sabatine, M.S. Genetic variants in ABCB1 and CYP2C19 and cardiovascular outcomes after treatment with clopidogrel and prasugrel in the TRITON-TIMI 38 trial: A pharmacogenetic analysis. Lancet 2010, 376, 1312–1319. [Google Scholar] [CrossRef]
- Tang, X.F.; Zhang, J.H.; Wang, J.; Han, Y.L.; Xu, B.; Qiao, S.B.; Wu, Y.J.; Chen, J.; Wu, Y.; Chen, J.L.; et al. Effects of coexisting polymorphisms of CYP2C19 and P2Y12 on clopidogrel responsiveness and clinical outcome in patients with acute coronary syndromes undergoing stent-based coronary intervention. Chin. Med. J. 2013, 126, 1069–1075. [Google Scholar] [PubMed]
- Shalia, K.K.; Shah, V.K.; Pawar, P.; Divekar, S.S.; Payannavar, S. Polymorphisms of MDR1, CYP2C19 and P2Y12 genes in Indian population: Effects on clopidogrel response. Indian Heart J. 2013, 65, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.A.; Sandhu, N.; Herrmann, J. Systems biology approaches to adverse drug effects: The example of cardio-oncology. Nat. Rev. Clin. Oncol. 2015, 12, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Mega, J.L.; Simon, T.; Collet, J.P.; Anderson, J.L.; Antman, E.M.; Bliden, K.; Cannon, C.P.; Danchin, N.; Giusti, B.; Gurbel, P.; et al. Reduced-function CYP2C19 genotype and risk of adverse clinical outcomes among patients treated with clopidogrel predominantly for PCI: A meta-analysis. JAMA 2010, 304, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.V.; Perel, P.; Shah, T.; Hingorani, A.D.; Casas, J.P. CYP2C19 genotype, clopidogrel metabolism, platelet function, and cardiovascular events: A systematic review and meta-analysis. JAMA 2011, 306, 2704–2714. [Google Scholar] [CrossRef] [PubMed]
- Marume, K.; Hokimoto, S.; Tabata, N.; Akasaka, T.; Tsujita, K.; Sakamoto, K.; Yamamoto, E.; Yamamuro, M.; Kaikita, K.; Oniki, K.; et al. Intraprocedural thrombotic event during coronary intervention depends on CYP2C19 genotype and is a predictor of future clinical event. Int. J. Cardiol. 2015, 187, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Martínez, R.; Fernández-Novoa, L.; Carril, J.C.; Lombardi, V.; Carrera, I.; Corzo, L.; Tellado, I.; Leszek, J.; McKay, A.; et al. Genomics of Dementia: APOE- and CYP2D6-Related Pharmacogenetics. Int. J. Alzheimers Dis. 2012, 2012, 518901. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Martinez-Bouza, R.; Carril, J.C.; Fernandez-Novoa, L.; Lombardi, V.; Carrera, I.; Corzo, L.; McKay, A. Genomics and pharmacogenomics of brain disorders. Curr. Pharm. Biotechnol. 2012, 13, 674–725. [Google Scholar] [CrossRef] [PubMed]
- Mani, H.; Toennes, S.W.; Linnemann, B.; Urbanek, D.A.; Schwonberg, J.; Kauert, G.F.; Lindhoff-Last, E. Determination of clopidogrel main metabolite in plasma: A useful tool for monitoring therapy? Ther. Drug Monit. 2008, 30, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Paniccia, R.; Priora, R.; Liotta, A.A.; Abbate, R. Platelet function tests: A comparative review. Vasc. Health Risk Manag. 2015, 11, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.H.; Bliden, K.P.; Antonino, M.J.; Park, K.S.; Tantry, U.S.; Gurbel, P.A. Usefulness of the VerifyNow P2Y12 assay to evaluate the antiplatelet effects of ticagrelor and clopidogrel therapies. Am Heart J. 2012, 164, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Gurbel, P.A.; Tantry, U.S. Drug insight: Clopidogrel nonresponsiveness. Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Gurbel, P.A.; Tantry, U.S. Clopidogrel resistance? Thromb. Res. 2007, 120, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Matetzky, S.; Shenkman, B.; Guetta, V.; Shechter, M.; Beinart, R.; Bienart, R.; Goldenberg, I.; Novikov, I.; Pres, H.; Savion, N.; et al. Clopidogrel resistance is associated with increased risk of recurrent atherothrombotic events in patients with acute myocardial infarction. Circulation 2004, 109, 3171–3175. [Google Scholar] [CrossRef] [PubMed]
- Geisler, T.; Langer, H.; Wydymus, M.; Göhring, K.; Zürn, C.; Bigalke, B.; Stellos, K.; May, A.E.; Gawaz, M. Low response to clopidogrel is associated with cardiovascular outcome after coronary stent implantation. Eur. Heart J. 2006, 27, 2420–2425. [Google Scholar] [CrossRef] [PubMed]
- Vavuranakis, M.; Vrachatis, D.A.; Papaioannou, T.G.; Archontakis, S.; Kalogeras, K.I.; Kariori, M.G.; Gafou, A.; Moldovan, C.; Tzamalis, P.; Stefanadis, C. Residual platelet reactivity after clopidogrel loading in patients with ST-elevation myocardial infarction undergoing an unexpectedly delayed primary percutaneous coronary intervention. -Impact on intracoronary thrombus burden and myocardial perfusion-. Circ. J. 2011, 75, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Dahabreh, I.; Moorthy, D.; Lamont, J.; Chen, M.; Kent, D.; Lau, J. Testing of CYP2C19 Variants and Platelet Reactivity for Guiding Antiplatelet Treatment; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2013.
- Siasos, G.; Oikonomou, E.; Vavuranakis, M.; Kokkou, E.; Mourouzis, K.; Tsalamandris, S.; Zaromitidou, M.; Kioufis, S.; Tsigkou, V.; Deftereos, S.; et al. Genotyping, Platelet Activation, and Cardiovascular Outcome in Patients after Percutaneous Coronary Intervention: Two Pieces of the Puzzle of Clopidogrel Resistance. Cardiology 2017, 137, 104–113. [Google Scholar] [CrossRef] [PubMed]
- L’Allier, P.L.; Ducrocq, G.; Pranno, N.; Noble, S.; Ibrahim, R.; Grégoire, J.C.; Azzari, F.; Nozza, A.; Berry, C.; Doucet, S.; et al. Clopidogrel 600-mg double loading dose achieves stronger platelet inhibition than conventional regimens: Results from the PREPAIR randomized study. J. Am. Coll. Cardiol. 2008, 51, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Buckland, R.; Daly, M.; Storey, R. Inhibition of platelet aggregation by clopidogrel is unaffected by the CYP2C19 681G > A polymorphism in patients with coronary artery disease (abstr). J. Am. Coll. Cardiol. 2007, 49, 375A. [Google Scholar]
- Umemura, K.; Furuta, T.; Kondo, K. The common gene variants of CYP2C19 affect pharmacokinetics and pharmacodynamics in an active metabolite of clopidogrel in healthy subjects. J. Thromb. Haemost. 2008, 6, 1439–1441. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.P.; Cuisset, T.; Rangé, G.; Cayla, G.; Elhadad, S.; Pouillot, C.; Henry, P.; Motreff, P.; Carrié, D.; Boueri, Z.; et al. Bedside monitoring to adjust antiplatelet therapy for coronary stenting. N. Engl. J. Med. 2012, 367, 2100–2109. [Google Scholar] [CrossRef] [PubMed]
- Cayla, G.; Cuisset, T.; Silvain, J.; Leclercq, F.; Manzo-Silberman, S.; Saint-Etienne, C.; Delarche, N.; Bellemain-Appaix, N.; Grégoire Range, G.; El Mahmoud, R.; et al. Platelet function monitoring to adjust antiplatelet therapy in elderly patients stented for an acute coronary syndrome (ANTARCTIC): An open-label, blinded-endpoint, randomised controlled superiority trial. Lancet 2016, 388, 2015–2022. [Google Scholar] [CrossRef]
- Hochholzer, W.; Ruff, C.T.; Mesa, R.A.; Mattimore, J.F.; Cyr, J.F.; Lei, L.; Frelinger, A.L.; Michelson, A.D.; Berg, D.D.; Angiolillo, D.J.; et al. Variability of individual platelet reactivity over time in patients treated with clopidogrel: Insights from the ELEVATE-TIMI 56 trial. J. Am. Coll. Cardiol. 2014, 64, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.J.; Ferreiro, J.L.; Price, M.J.; Kirtane, A.J.; Stone, G.W. Platelet function and genetic testing. J. Am. Coll. Cardiol. 2013, 62, S21–S31. [Google Scholar] [CrossRef] [PubMed]
- Frelinger, A.L.; Bhatt, D.L.; Lee, R.D.; Mulford, D.J.; Wu, J.; Nudurupati, S.; Nigam, A.; Lampa, M.; Brooks, J.K.; Barnard, M.R.; et al. Clopidogrel pharmacokinetics and pharmacodynamics vary widely despite exclusion or control of polymorphisms (CYP2C19, ABCB1, PON1), noncompliance, diet, smoking, co-medications (including proton pump inhibitors), and pre-existent variability in platelet function. J. Am. Coll. Cardiol. 2013, 61, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.T.; Payne, C.D.; Wiviott, S.D.; Weerakkody, G.; Farid, N.A.; Small, D.S.; Jakubowski, J.A.; Naganuma, H.; Winters, K.J. A comparison of prasugrel and clopidogrel loading doses on platelet function: Magnitude of platelet inhibition is related to active metabolite formation. Am. Heart J. 2007, 153, 66.e9–66.e16. [Google Scholar] [CrossRef] [PubMed]
- Gong, I.Y.; Crown, N.; Suen, C.M.; Schwarz, U.I.; Dresser, G.K.; Knauer, M.J.; Sugiyama, D.; DeGorter, M.K.; Woolsey, S.; Tirona, R.G.; et al. Clarifying the importance of CYP2C19 and PON1 in the mechanism of clopidogrel bioactivation and in vivo antiplatelet response. Eur. Heart J. 2012, 33, 2856–2864. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.K.; Subbaiah, G.; Shah, H.; Kundlik, M.; Shrivastav, P.S. Rapid LC-ESI-MS-MS method for the simultaneous determination of clopidogrel and its carboxylic acid metabolite in human plasma. J. Chromatogr. Sci. 2008, 46, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yin, T.; Li, Y.; Song, L.Q.; Yu, J.; Si, R.; Zhang, Y.M.; He, Y.; Guo, W.Y.; Wang, H.C. CYP2C19 polymorphisms and coronary heart disease risk factors synergistically impact clopidogrel response variety after percutaneous coronary intervention. Coron. Artery Dis. 2014, 25, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Chen, M.; Liu, X.J.; Liu, W.; Li, Q.; Chai, H.; Ren, X.; Wang, X.Q.; Zhao, Z.G.; Zhang, C.; et al. The CYP2C19 genotype does not impact the long-term prognosis of patients with coronary artery disease. Atherosclerosis 2013, 227, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Paré, G.; Eikelboom, J.W.; Simonsen, K.L.; Emison, E.S.; Fox, K.A.; Steg, P.G.; Montalescot, G.; Bhakta, N.; Hacke, W.; et al. The relationship between CYP2C19 polymorphisms and ischaemic and bleeding outcomes in stable outpatients: The CHARISMA genetics study. Eur. Heart J. 2012, 33, 2143–2150. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.J.; Pogue, J.; Hart, R.G.; Hohnloser, S.H.; Pfeffer, M.; Chrolavicius, S.; Yusuf, S.; Investigators, A. Effect of clopidogrel added to aspirin in patients with atrial fibrillation. N. Engl. J. Med. 2009, 360, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Pereira, N.L.; Sargent, D.J.; Farkouh, M.E.; Rihal, C.S. Genotype-based clinical trials in cardiovascular disease. Nat. Rev. Cardiol. 2015, 12, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Tailored Antiplatelet Therapy Following PCI (TAILOR-PCI). 2015. Available online: https://www.clinicaltrials.gov/ct2/show/NCT01742117 (accessed on 20 September 2017).
- Paré, G.; Mehta, S.R.; Yusuf, S.; Anand, S.S.; Connolly, S.J.; Hirsh, J.; Simonsen, K.; Bhatt, D.L.; Fox, K.A.; Eikelboom, J.W. Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N. Engl. J. Med. 2010, 363, 1704–1714. [Google Scholar] [CrossRef] [PubMed]
- Hochholzer, W.; Trenk, D.; Fromm, M.F.; Valina, C.M.; Stratz, C.; Bestehorn, H.P.; Büttner, H.J.; Neumann, F.J. Impact of cytochrome P450 2C19 loss-of-function polymorphism and of major demographic characteristics on residual platelet function after loading and maintenance treatment with clopidogrel in patients undergoing elective coronary stent placement. J. Am. Coll. Cardiol. 2010, 55, 2427–2434. [Google Scholar] [CrossRef] [PubMed]
- Gachet, C.; Aleil, B. Testing antiplatelet therapy. Eur. Heart J. Suppl. 2008, 10, A28–A34. [Google Scholar] [CrossRef]
- Kim, Y.G.; Suh, J.W.; Park, J.J.; Oh, I.Y.; Yoon, C.H.; Cho, Y.S.; Youn, T.J.; Chae, I.H.; Choi, D.J. Different influences of hematocrit on the results of two Point-Of-Care platelet function tests, the VerifyNow assay and multiple electrode platelet aggregometry. PLoS ONE 2014, 9, e114053. [Google Scholar] [CrossRef] [PubMed]
- Kakouros, N.; Kickler, T.S.; Laws, K.M.; Rade, J.J. Hematocrit alters VerifyNow P2Y12 assay results independently of intrinsic platelet reactivity and clopidogrel responsiveness. J. Thromb. Haemost. 2013, 11, 1814–1822. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.; Cheng-Lai, A.; Nawarskas, J. Clopidogrel and genetic testing: Is it necessary for everyone? Cardiol. Rev. 2012, 20, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Kirtane, A.J.; Stone, G.W. How to minimize stent thrombosis. Circulation 2011, 124, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Bhatt, D.L.; Brennan, D.M.; Hankey, G.J.; Steinhubl, S.R.; Johnston, S.C.; Montalescot, G.; Mak, K.H.; Fox, K.A.; Easton, D.J.; et al. High-sensitivity C-reactive protein and clopidogrel treatment in patients at high risk of cardiovascular events: A substudy from the CHARISMA trial. Heart. 2011, 97, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Shi, D.; Yang, D.; Song, X.; Yan, B. Interleukin-6 alters the cellular responsiveness to clopidogrel, irinotecan, and oseltamivir by suppressing the expression of carboxylesterases HCE1 and HCE2. Mol. Pharmacol. 2007, 72, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Hajsadeghi, S.; Chitsazan, M.; Chitsazan, M.; Salehi, N.; Amin, A.; Maleki, M.; Babaali, N.; Abdi, S.; Mohsenian, M. Changes of High Sensitivity C-Reactive Protein During Clopidogrel Therapy in Patients Undergoing Percutaneous Coronary Intervention. Res. Cardiovasc. Med. 2016, 5, e28997. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.J.; Ferreiro, J.L. Platelet adenosine diphosphate P2Y12 receptor antagonism: Benefits and limitations of current treatment strategies and future directions. Rev. Esp. Cardiol. 2010, 63, 60–76. [Google Scholar] [CrossRef]
- Desta, Z.; Zhao, X.; Shin, J.G.; Flockhart, D.A. Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin. Pharmacokinet. 2002, 41, 913–958. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.R.; Poland, R.E.; Lin, K.M.; Wan, Y.J. Genetic polymorphism of cytochrome P450 2C19 in Mexican Americans: A cross-ethnic comparative study. Clin. Pharmacol. Ther. 2006, 80, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, A.; Kaneko, O.; Taleo, G.; Björkman, A.; Kobayakawa, T. High frequencies of CYP2C19 mutations and poor metabolism of proguanil in Vanuatu. Lancet 1997, 349, 921–922. [Google Scholar] [CrossRef]
- Hsu, H.L.; Woad, K.J.; Woodfield, D.G.; Helsby, N.A. A high incidence of polymorphic CYP2C19 variants in archival blood samples from Papua New Guinea. Hum. Genom. 2008, 3, 17–23. [Google Scholar] [CrossRef]
- Li-Wan-Po, A.; Girard, T.; Farndon, P.; Cooley, C.; Lithgow, J. Pharmacogenetics of CYP2C19: Functional and clinical implications of a new variant CYP2C19*17. Br. J. Clin. Pharmacol. 2010, 69, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, L.K.; Torguson, R.; Loh, J.P.; Devaney, J.M.; Chen, F.; Kitabata, H.; Minha, S.; Barbash, I.M.; Suddath, W.O.; Satler, L.F.; et al. Racial disparity with on-treatment platelet reactivity in patients undergoing percutaneous coronary intervention. Am. Heart J. 2013, 166, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Tracy, T.S.; Chaudhry, A.S.; Prasad, B.; Thummel, K.E.; Schuetz, E.G.; Zhong, X.B.; Tien, Y.C.; Jeong, H.; Pan, X.; Shireman, L.M. Interindividual Variability in Cytochrome P450-Mediated Drug Metabolism. Drug Metab. Dispos. 2016, 44, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Giri, A.K.; Khan, N.M.; Basu, A.; Tandon, N.; Scaria, V.; Bharadwaj, D. Pharmacogenetic landscape of clopidogrel in north Indians suggest distinct interpopulation differences in allele frequencies. Pharmacogenomics 2014, 15, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Shetkar, S.S.; Ramakrishnan, S.; Seth, S.; Chandna, P.; Verma, S.K.; Bhargava, B.; Bahl, V.K. CYP 450 2C19 polymorphisms in Indian patients with coronary artery disease. Indian Heart J. 2014, 66, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.S.; Cho, K.I.; Jin, H.Y.; Seo, J.S.; Yang, T.H.; Kim, D.K.; Kim, D.S.; Seol, S.H.; Kim, D.I.; Kim, B.H.; et al. Meta-analysis of cytochrome P450 2C19 polymorphism and risk of adverse clinical outcomes among coronary artery disease patients of different ethnic groups treated with clopidogrel. Am. J. Cardiol. 2012, 110, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Hokimoto, S.; Mizobe, M.; Akasaka, T.; Arima, Y.; Kaikita, K.; Nakagawa, K.; Ogawa, H. Impact of CYP2C19 polymorphism and proton pump inhibitors on platelet reactivity to clopidogrel and clinical outcomes following stent implantation. Thromb. Res. 2014, 133, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Chang, K.; Koh, Y.S.; Park, M.W.; Choi, Y.S.; Park, C.S.; Oh, M.; Kim, E.Y.; Shon, J.H.; Shin, J.G.; et al. CYP2C19 poor metabolizer is associated with clinical outcome of clopidogrel therapy in acute myocardial infarction but not stable angina. Circ. Cardiovasc. Genet. 2013, 6, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.H.; Tantry, U.S.; Kim, I.S.; Koh, J.S.; Kwon, T.J.; Park, Y.; Hwang, S.J.; Bliden, K.P.; Kwak, C.H.; Hwang, J.Y.; et al. Effect of CYP2C19*2 and *3 loss-of-function alleles on platelet reactivity and adverse clinical events in East Asian acute myocardial infarction survivors treated with clopidogrel and aspirin. Circ. Cardiovasc. Interv. 2011, 4, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Pharmacogene Variation (PharmVar) Consortium. 2017. Available online: https://www.pharmvar.org/ (accessed on 20 September 2017).
- Bates, E.R.; Lau, W.C.; Angiolillo, D.J. Clopidogrel-drug interactions. J. Am. Coll. Cardiol. 2011, 57, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.S.; Hlatky, M.A.; Antman, E.M.; Bhatt, D.L.; Bjorkman, D.J.; Clark, C.B.; Furberg, C.D.; Johnson, D.A.; Kahi, C.J.; Laine, L.; et al. ACCF/ACG/AHA 2010 expert consensus document on the concomitant use of proton pump inhibitors and thienopyridines: A focused update of the ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use. A Report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents. J. Am. Coll. Cardiol. 2010, 56, 2051–2066. [Google Scholar] [CrossRef] [PubMed]
- Siller-Matula, J.M.; Spiel, A.O.; Lang, I.M.; Kreiner, G.; Christ, G.; Jilma, B. Effects of pantoprazole and esomeprazole on platelet inhibition by clopidogrel. Am. Heart J. 2009, 157, 148.e1–148.e5. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Andersson, T.B.; Ahlström, M.; Weidolf, L. Comparison of inhibitory effects of the proton pump-inhibiting drugs omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole on human cytochrome P450 activities. Drug Metab. Dispos. 2004, 32, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Serebruany, V.; Cherala, G.; Williams, C.; Surigin, S.; Booze, C.; Kuliczkowski, W.; Atar, D. Association of platelet responsiveness with clopidogrel metabolism: Role of compliance in the assessment of “resistance”. Am. Heart J. 2009, 158, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Geisler, T.; Schaeffeler, E.; Dippon, J.; Winter, S.; Buse, V.; Bischofs, C.; Zuern, C.; Moerike, K.; Gawaz, M.; Schwab, M. CYP2C19 and nongenetic factors predict poor responsiveness to clopidogrel loading dose after coronary stent implantation. Pharmacogenomics 2008, 9, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.J.; Fernández-Ortiz, A.; Bernardo, E.; Barrera Ramírez, C.; Sabaté, M.; Fernandez, C.; Hernández-Antolín, R.; Escaned, J.; Alfonso, F.; Macaya, C. Platelet aggregation according to body mass index in patients undergoing coronary stenting: Should clopidogrel loading-dose be weight adjusted? J. Invasive Cardiol. 2004, 16, 169–174. [Google Scholar] [PubMed]
- Holmberg, M.T.; Tornio, A.; Neuvonen, M.; Neuvonen, P.J.; Backman, J.T.; Niemi, M. Grapefruit juice inhibits the metabolic activation of clopidogrel. Clin. Pharmacol. Ther. 2014, 95, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Lev, E.I.; Arikan, M.E.; Vaduganathan, M.; Alviar, C.L.; Tellez, A.; Mathuria, N.; Builes, A.; Granada, J.F.; del Conde, I.; Kleiman, N.S. Effect of caffeine on platelet inhibition by clopidogrel in healthy subjects and patients with coronary artery disease. Am. Heart J. 2007, 154, 694.e1–694.e7. [Google Scholar] [CrossRef] [PubMed]
- Bliden, K.P.; Dichiara, J.; Lawal, L.; Singla, A.; Antonino, M.J.; Baker, B.A.; Bailey, W.L.; Tantry, U.S.; Gurbel, P.A. The association of cigarette smoking with enhanced platelet inhibition by clopidogrel. J. Am. Coll. Cardiol. 2008, 52, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.J.; Bernardo, E.; Zanoni, M.; Vivas, D.; Capranzano, P.; Malerba, G.; Capodanno, D.; Prandini, P.; Pasquali, A.; Trabetti, E.; et al. Impact of insulin receptor substrate-1 genotypes on platelet reactivity and cardiovascular outcomes in patients with type 2 diabetes mellitus and coronary artery disease. J. Am. Coll. Cardiol. 2011, 58, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.J.; Fernandez-Ortiz, A.; Bernardo, E.; Ramírez, C.; Sabaté, M.; Jimenez-Quevedo, P.; Hernández, R.; Moreno, R.; Escaned, J.; Alfonso, F.; et al. Platelet function profiles in patients with type 2 diabetes and coronary artery disease on combined aspirin and clopidogrel treatment. Diabetes 2005, 54, 2430–2435. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Braunwald, E.; Angiolillo, D.J.; Meisel, S.; Dalby, A.J.; Verheugt, F.W.; Goodman, S.G.; Corbalan, R.; Purdy, D.A.; Murphy, S.A.; et al. Greater clinical benefit of more intensive oral antiplatelet therapy with prasugrel in patients with diabetes mellitus in the trial to assess improvement in therapeutic outcomes by optimizing platelet inhibition with prasugrel-Thrombolysis in Myocardial Infarction 38. Circulation 2008, 118, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Caillard, S.; Jesel, L.; El Ghannudi, S.; Ohlmann, P.; Sauleau, E.; Hannedouche, T.; Gachet, C.; Moulin, B.; Morel, O. Association of estimated GFR with platelet inhibition in patients treated with clopidogrel. Am. J. Kidney Dis. 2012, 59, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Gremmel, T.; Müller, M.; Steiner, S.; Seidinger, D.; Koppensteiner, R.; Kopp, C.W.; Panzer, S. Chronic kidney disease is associated with increased platelet activation and poor response to antiplatelet therapy. Nephrol. Dial. Transplant. 2013, 28, 2116–2122. [Google Scholar] [CrossRef] [PubMed]
- Geisler, T.; Grass, D.; Bigalke, B.; Stellos, K.; Drosch, T.; Dietz, K.; Herdeg, C.; Gawaz, M. The Residual Platelet Aggregation after Deployment of Intracoronary Stent (PREDICT) score. J. Thromb. Haemost. 2008, 6, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Best, P.J.; Lennon, R.; Ting, H.H.; Bell, M.R.; Rihal, C.S.; Holmes, D.R.; Berger, P.B. The impact of renal insufficiency on clinical outcomes in patients undergoing percutaneous coronary interventions. J. Am. Coll. Cardiol. 2002, 39, 1113–1119. [Google Scholar] [CrossRef]
- Angiolillo, D.J.; Bernardo, E.; Capodanno, D.; Vivas, D.; Sabaté, M.; Ferreiro, J.L.; Ueno, M.; Jimenez-Quevedo, P.; Alfonso, F.; Bass, T.A.; et al. Impact of chronic kidney disease on platelet function profiles in diabetes mellitus patients with coronary artery disease taking dual antiplatelet therapy. J. Am. Coll. Cardiol. 2010, 55, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.E.; Larson, M.G.; Tanriverdi, K.; O’Donnell, C.J.; Morin, K.; Hakanson, A.S.; Vasan, R.S.; Johnson, A.D.; Iafrati, M.D.; Benjamin, E.J. Relation of platelet and leukocyte inflammatory transcripts to body mass index in the Framingham heart study. Circulation 2010, 122, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.L.; Bhargava, P.; Cherrouk, I.; Marshall, J.L.; Flockhart, D.A.; Wainer, I.W. A discordance of the cytochrome P450 2C19 genotype and phenotype in patients with advanced cancer. Br. J. Clin. Pharmacol. 2000, 49, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.F.; Schneider, V.M.; Frye, C.S.; Feldman, A.M. Plasma levels of TNF-alpha and IL-6 are inversely related to cytochrome P450-dependent drug metabolism in patients with congestive heart failure. J. Card. Fail. 2002, 8, 315–319. [Google Scholar] [CrossRef] [PubMed]
- McGready, R.; Stepniewska, K.; Seaton, E.; Cho, T.; Cho, D.; Ginsberg, A.; Edstein, M.D.; Ashley, E.; Looareesuwan, S.; White, N.J.; et al. Pregnancy and use of oral contraceptives reduces the biotransformation of proguanil to cycloguanil. Eur J. Clin. Pharmacol. 2003, 59, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, Y.; Yasui-Furukori, N.; Takahata, T.; Sasaki, M.; Tateishi, T. The effect of aging on the relationship between the cytochrome P450 2C19 genotype and omeprazole pharmacokinetics. Clin. Pharmacokinet. 2005, 44, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Habano, W.; Kawamura, K.; Iizuka, N.; Terashima, J.; Sugai, T.; Ozawa, S. Analysis of DNA methylation landscape reveals the roles of DNA methylation in the regulation of drug metabolizing enzymes. Clin. Epigenet. 2015, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Li, X.; Yu, Q.; Liu, Y.; Wang, Y.; Song, H.; Cui, H.; Du, W.; Fei, X.; Liu, J.; et al. Association of P2Y12 gene promoter DNA methylation with the risk of clopidogrel resistance in coronary artery disease patients. Biomed. Res. Int. 2014, 2014, 450814. [Google Scholar] [CrossRef] [PubMed]
- Li, X.G.; Ma, N.; Wang, B.; Li, X.Q.; Mei, S.H.; Zhao, K.; Wang, Y.J.; Li, W.; Zhao, Z.G.; Sun, S.S.; et al. The impact of P2Y12 promoter DNA methylation on the recurrence of ischemic events in Chinese patients with ischemic cerebrovascular disease. Sci. Rep. 2016, 6, 34570. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010, 11, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M.; Frost, R.J.; Olson, E.N. MicroRNAs add a new dimension to cardiovascular disease. Circulation 2010, 121, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, A.; Skaar, T.C. In silico identification of microRNAs predicted to regulate the drug metabolizing cytochrome P450 genes. Drug Metab. Lett. 2011, 5, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Surapureddi, S.; Coulter, S.; Ferguson, S.S.; Goldstein, J.A. Human CYP2C8 is post-transcriptionally regulated by microRNAs 103 and 107 in human liver. Mol. Pharmacol. 2012, 82, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Caruso, R.; Rocchiccioli, S.; Gori, A.M.; Cecchettini, A.; Giusti, B.; Parodi, G.; Cozzi, L.; Marcucci, R.; Parolini, M.; Romagnuolo, I.; et al. Inflammatory and antioxidant pattern unbalance in “clopidogrel-resistant” patients during acute coronary syndrome. Mediat. Inflamm. 2015, 2015, 710123. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, R. Metabolomics of a superorganism. J. Nutr. 2007, 137, 259S–266S. [Google Scholar] [CrossRef] [PubMed]
- Semmar, N. Metabotype concept: Flexibility, usefulness and meaning in different biological populations. In Metabolomics; Roessner, D., Ed.; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Velasquez, M.T.; Ramezani, A.; Manal, A.; Raj, D.S. Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins 2016, 8, 326. [Google Scholar] [CrossRef] [PubMed]
- Senthong, V.; Wang, Z.; Li, X.S.; Fan, Y.; Wu, Y.; Tang, W.H.; Hazen, S.L. Intestinal Microbiota-Generated Metabolite Trimethylamine-N-Oxide and 5-Year Mortality Risk in Stable Coronary Artery Disease: The Contributory Role of Intestinal Microbiota in a COURAGE-Like Patient Cohort. J. Am. Heart Assoc. 2016, 5, 178.e1–178.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wang, Z.; Tang, W.H.W.; Hazen, S.L. Gut Microbe-Generated Trimethylamine N-Oxide From Dietary Choline Is Prothrombotic in Subjects. Circulation 2017, 135, 1671–1673. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.M.; Aronoff, D.M. The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin. Microbiol. Infect. 2016, 22. [Google Scholar] [CrossRef] [PubMed]
- Djebli, N.; Fabre, D.; Boulenc, X.; Fabre, G.; Sultan, E.; Hurbin, F. Physiologically based pharmacokinetic modeling for sequential metabolism: Effect of CYP2C19 genetic polymorphism on clopidogrel and clopidogrel active metabolite pharmacokinetics. Drug Metab. Dispos. 2015, 43, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Saab, Y.B.; Zeenny, R.; Ramadan, W.H. Optimizing clopidogrel dose response: A new clinical algorithm comprising CYP2C19 pharmacogenetics and drug interactions. Ther. Clin. Risk Manag. 2015, 11, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.J.; Fernandez-Ortiz, A.; Bernardo, E.; Ramírez, C.; Cavallari, U.; Trabetti, E.; Sabaté, M.; Hernández, R.; Moreno, R.; Escaned, J.; et al. Contribution of gene sequence variations of the hepatic cytochrome P450 3A4 enzyme to variability in individual responsiveness to clopidogrel. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1895–1900. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Miyata, T. Pharmacogenomics of clopidogrel: Evidence and perspectives. Thromb. Res. 2011, 128, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Price, M.J.; Murray, S.S.; Angiolillo, D.J.; Lillie, E.; Smith, E.N.; Tisch, R.L.; Schork, N.J.; Teirstein, P.S.; Topol, E.J.; Investigators, G. Influence of genetic polymorphisms on the effect of high- and standard-dose clopidogrel after percutaneous coronary intervention: The GIFT (Genotype Information and Functional Testing) study. J. Am. Coll. Cardiol. 2012, 59, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Mega, J.L.; Hochholzer, W.; Frelinger, A.L.; Kluk, M.J.; Angiolillo, D.J.; Kereiakes, D.J.; Isserman, S.; Rogers, W.J.; Ruff, C.T.; Contant, C.; et al. Dosing clopidogrel based on CYP2C19 genotype and the effect on platelet reactivity in patients with stable cardiovascular disease. JAMA 2011, 306, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Pereira, N.L.; Stewart, A.K. Clinical Implementation of Cardiovascular Pharmacogenomics. Mayo Clin. Proc. 2015, 90, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Amsterdam, E.A.; Wenger, N.K.; Brindis, R.G.; Casey, D.E.; Ganiats, T.G.; Holmes, D.R.; Jaffe, A.S.; Jneid, H.; Kelly, R.F.; Kontos, M.C.; et al. 2014 AHA/ACC Guideline for the Management of Patients with Non-ST-Elevation Acute Coronary Syndromes: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2014, 64, e139–e228. [Google Scholar] [CrossRef] [PubMed]
- Cost-Effectiveness of Genotype Guided Treatment with Antiplatelet Drugs in Stemi Patients: Optimization of Treatment (Popgenetics). 2013. Available online: https://www.clinicaltrials.gov/ct2/show/NCT01761786 (accessed on 20 September 2017).
- Offit, K. Personalized medicine: New genomics, old lessons. Hum. Genet. 2011, 130, 3–14. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute: Personalized Medicine. Dictionary of Cancer Terms. 2016. Available online: http://www.cancer.gov/publications/dictionaries/cancer-terms?cdrid=561717 (accessed on 20 September 2017).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Allele | Characteristic SNP a | Functional Change | References | ||
|---|---|---|---|---|---|
| cDNA | Gene | Effect | |||
| CYP2C19*1 | None 1 | None | None | Normal | [49] |
| CYP2C19*2 | 681G>A 2 | 19154G>A | Splicing defect | Non-functional | [50,51,52,53,54] |
| CYP2C19*3 | 636G>A 3 | 17948G>A | Premature stop codon (W212X) | Non-functional | [52,55] |
| CYP2C19*4 | 1A>G 4 | 1A>G | GTG initiation codon | Non-functional | [56,57] |
| CYP2C19*5 | 1297C>T 5 | 90033C>T | R433W | Non-functional | [58,59] |
| CYP2C19*6 | 395G>A | 12748G>A | R132O | Non-functional | [51] |
| CYP2C19*7 | 19294T>A | Splicing defect | Non-functional | [60] | |
| CYP2C19*8 | 358T>C | 12711T>C | W120R | Decreased in vitro | [60] |
| CYP2C19*9 | 431G>A | 12784G>A | R144H | Decreased in vitro | [61] |
| CYP2C19*10 | 680C>T | 19153C>T | P227L | Decreased in vitro | [61] |
| CYP2C19*11 | 449G>A | 12802G>A | R150H | Similar to wild type in vitro | [61] |
| CYP2C19*12 | 1473A>C | 90209A>C | X491C; 26 extra amino acids | Unstable in vitro | [61] |
| CYP2C19*13 | 1228C>T | 87290C>T | R410C | Similar to wild type in vitro | [61] |
| CYP2C19*14 | 50T>C | 50T> C | L17P | Not determined | [61] |
| CYP2C19*15 | 55A>C | 55A>C | I19L | Not determined | [61] |
| CYP2C19*16 | 1324C>T 6 | 90060C>T | R442C | Not determined | [62] |
| CYP2C19*17 | 3402C>T | Increased transcription in vitro, should not be termed Ultrarapid (UM) | [63] | ||
| −806C>T | |||||
| CYP2C19*18 | 986G>A | 80156G>A | R329H | Not determined | [51] |
| 87106T>C | |||||
| CYP2C19*19 | 151A>G | 151A>G | S51G | Not determined | [51] |
| 87106T>C | |||||
| CYP2C19*20 7 | 636G>A | 17948G>A | Premature stop codon (W212X) and D360N | Non-functional | [51] |
| CYP2C19*21 8 | 681G>A | 19154G>A | Splicing defect and A161P | Non-functional | [51,54] |
| −98T>C | |||||
| CYP2C19*22 | 557G>C | 17869G>C | R186P and G91R | Not determined | [64] |
| CYP2C19*23 | 271G>C | 12455G> C | R335O | Not determined | [65] |
| CYP2C19*24 | 1004G>A | 80174G>A | F448L | Not determined | [65] |
| 1197A>G | 87259A>G | ||||
| CYP2C19*25 | 1344C>G | 90080C>G | D256N | Not determined | [65] |
| CYP2C19*26 | 766G>A | 19239G>A | V374I | Decreased in vitro | [53] |
| CYP2C19*27 | −1041G>A | Decreased in vitro | [66] | ||
| CYP2C19*28 | 1120G>A | −2020C>A | No significant decrease in vitro | [66] | |
| −1439T>C | |||||
| 80290G>A | |||||
| Phenotype | Example Genotypes | Enzyme Activity |
|---|---|---|
| Ultra-rapid metabolizer (UM) | *1/*17 | Normal or increased |
| *17/*17 | ||
| Extensive metabolizer (EM) | *1/*1 (wild type) | Normal |
| Intermediate metabolizer (IM) | *1/*2 | Intermediate |
| *1/*3 | ||
| *2/*17 | Likely intermediate | |
| *3/17 | Likely intermediate | |
| Poor metabolizer (PM) | *2/*2 | Low or absent |
| *3/*3 | ||
| *2/*3 |
| Allele | Defining Variants | Variant Type | Allele Frequencies in Indicated Populations, % | Functional Consequence | ||||
|---|---|---|---|---|---|---|---|---|
| EUR | AFR | EAS | SAS | AMR | ||||
| *1 | None | 59.2 | 44.5 | 60.5 | 51.9 | 77 | ||
| *2 | rs4244285 | Splicing defect | 18.3 | 18.1 | 31 | 34 | 10.1 | Inactive |
| *3 | rs4986893 | Stop-gain (W212X) | <0.1 | <0.1 | 6.7 | 0.4 | <0.1 | Inactive |
| *4 | rs28399504 | Start lost | 0 | <0.1 | <0.1 | <0.1 | 0.2 | Inactive |
| *5 | rs56337013 | Missense (R433W) | 0 | 0 | 0 | <0.1 | 0 | Inactive |
| *6 | rs72552267 | Missense (R132Q) | 0 | 0 | <0.1 | 0 | <0.1 | Inactive |
| *7 | rs72558186 | Splicing defect | 0 | 0 | 0 | <0.1 | 0 | Inactive b |
| *8 | rs41291556 | Missense (W120R) | <0.1 | <0.1 | 0 | <0.1 | <0.1 | Inactive |
| *9 | rs17884712 | Missense (R144H) | 0 | 1.2 | 0 | <0.1 | <0.1 | |
| *10 | rs6413438 | Missense (P227L) | 0 | 0.4 | <0.1 | 0 | <0.1 | Decreased a |
| *12 | rs55640102 | Stop-lost (X491C) | 0 | <0.1 | 0 | 0 | 0 | Decreased a |
| *13 | rs17879685 | Missense (R410C) | 0 | 1.6 | 0 | <0.1 | 0.1 | |
| *15 | rs17882687 | Missense (I19L) | 0 | 2 | 0 | <0.1 | <0.1 | |
| *16 | rs192154563 | Missense (R442C) | 0 | <0.1 | 0 | <0.1 | 0 | |
| *17 | rs12248560 | Regulatory | 22.4 | 23.5 | 1.5 | 13.6 | 12 | Increased |
| *22 | rs140278421 | Missense (R186P) | 0 | 0.1 | 0 | 0 | <0.1 | |
| *23 | rs118203756 | Missense (G91R) | 0 | 0 | <0.1 | 0 | 0 | |
| *24 | rs118203757 | Missense (R335Q) | 0 | <0.1 | 0 | <0.1 | <0.1 | |
| *25 | rs118203759 | Missense (F448L) | 0 | 0 | 0 | 0 | 0 | |
| *27 | rs7902257 | Regulatory | 0.1 | 8.3 | 0.1 | 0 | 0.3 | Decreased a |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, S.-A.; Pereira, N. Pharmacogenomic Impact of CYP2C19 Variation on Clopidogrel Therapy in Precision Cardiovascular Medicine. J. Pers. Med. 2018, 8, 8. https://doi.org/10.3390/jpm8010008
Brown S-A, Pereira N. Pharmacogenomic Impact of CYP2C19 Variation on Clopidogrel Therapy in Precision Cardiovascular Medicine. Journal of Personalized Medicine. 2018; 8(1):8. https://doi.org/10.3390/jpm8010008
Chicago/Turabian StyleBrown, Sherry-Ann, and Naveen Pereira. 2018. "Pharmacogenomic Impact of CYP2C19 Variation on Clopidogrel Therapy in Precision Cardiovascular Medicine" Journal of Personalized Medicine 8, no. 1: 8. https://doi.org/10.3390/jpm8010008