Pharmacogenomics of CYP2C9: Functional and Clinical Considerations †


Abstract
:1. Introduction
2. CYP2C9 Substrate Selectivity
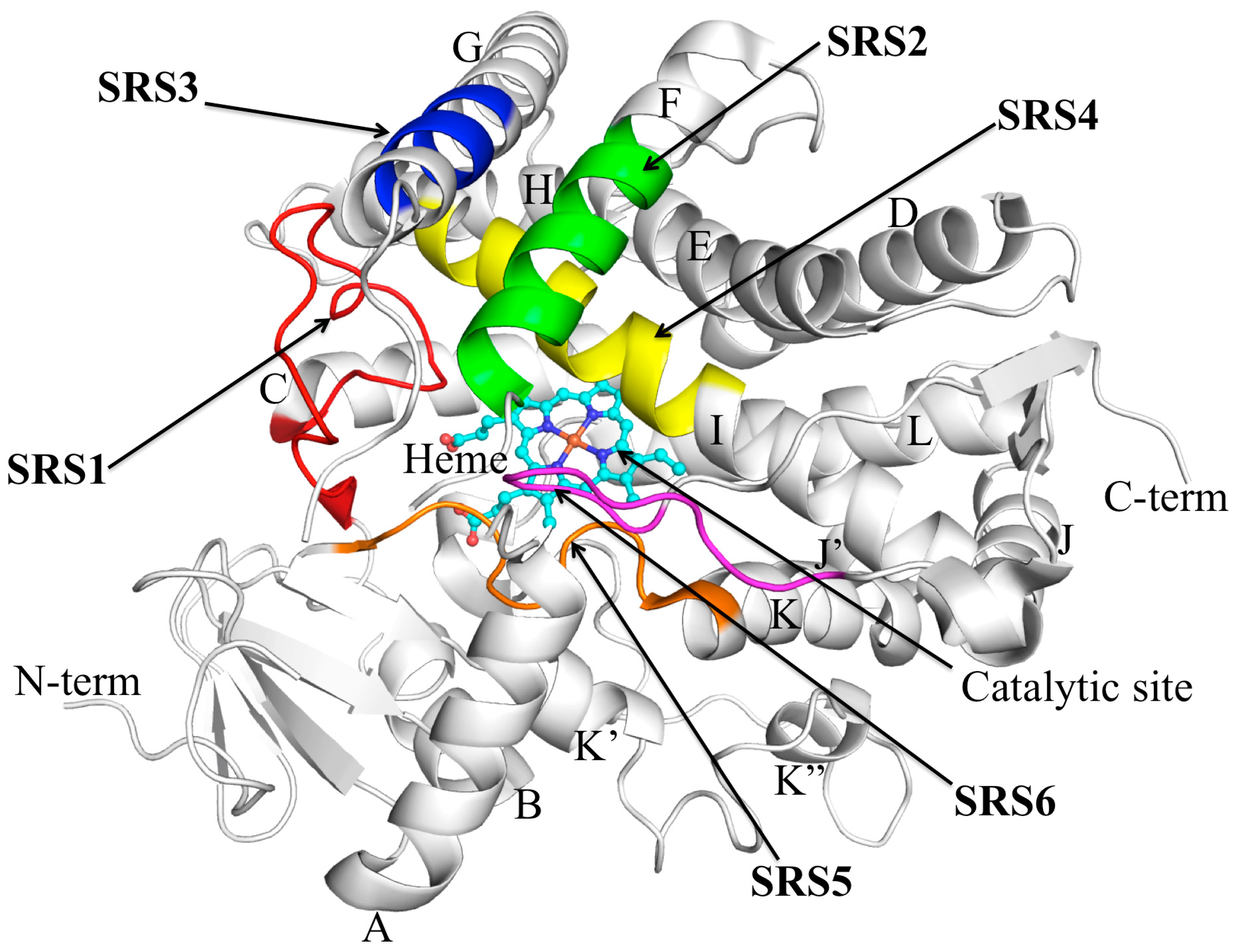
3. CYP2C9 Structure–Function
4. Clinical Relevance of CYP2C9
4.1. Clinically Relevant Substrates
4.2. CYP2C9 Inducers and Inhibitors
5. CYP2C9 Genetic Polymorphisms
5.1. Background
5.2. Missense and Frameshift Variants in CYP2C9
5.3. Functional Significance of CYP2C9 Missense Variants
5.4. Variants in Non-Coding Regions
5.5. Linkage Disequilibrium with Other CYP2C Genes
6. Clinical Significance of CYP2C9 Polymorphisms
6.1. Coumarin Anticoagulants
6.2. Sulfonylureas
6.3. Nonsteroidal Antiinflammatory Drugs
6.4. Phenytoin
6.5. Miscellaneous
7. Warfarin Dosing Algorithms
8. Contemporary Translational Efforts
8.1. Pre-Emptive Genotyping
8.2. The Problem of Variants of Uncertain Significance
9. Future Prospects
9.1. Computational Approaches
9.2. Large-Scale Functional Assays
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, H.F.; Wang, H.H.; Gao, N.; Wei, J.Y.; Tian, X.; Zhao, Y.; Fang, Y.; Zhou, J.; Wen, Q.; Gao, J.; et al. Physiological Content and Intrinsic Activities of 10 Cytochrome P450 Isoforms in Human Normal Liver Microsomes. J. Pharmacol. Exp. Ther. 2016, 358, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Paine, M.F.; Hart, H.L.; Ludington, S.S.; Haining, R.L.; Rettie, A.E.; Zeldin, D.C. The human intestinal cytochrome P450 “pie”. Drug Metab. Dispos. 2006, 34, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Isvoran, A.; Louet, M.; Vladoiu, D.L.; Craciun, D.; Loriot, M.A.; Villoutreix, B.O.; Miteva, M.A. Pharmacogenomics of the cytochrome P450 2C family: Impacts of amino acid variations on drug metabolism. Drug Discov. Today 2017, 22, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Veronese, M.E.; Miners, J.O.; Rees, D.L.; Birkett, D.J. Tolbutamide hydroxylation in humans: Lack of bimodality in 106 healthy subjects. Pharmacogenetics 1993, 3, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.O.; Birkett, D.J. Cytochrome P4502C9: An enzyme of major importance in human drug metabolism. Br. J. Clin. Pharmacol. 1998, 45, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Rettie, A.E.; Jones, J.P. Clinical and toxicological relevance of CYP2C9: Drug-Drug Interactions and Pharmacogenetics. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 477–494. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Zhou, Z.W.; Yang, L.P.; Cai, J.P. Substrates, inducers, inhibitors and structure-activity relationships of human Cytochrome P450 2C9 and implications in drug development. Curr. Med. Chem. 2009, 16, 3480–3675. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-F.; Zhou, Z.-W.; Huang, M. Polymorphisms of human cytochrome P450 2C9 and the functional relevance. Toxicology 2010, 278, 165–188. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, M. Genetic Polymorphisms and in Vitro Functional Characterization of CYP2C8, CYP2C9, and CYP2C19 Allelic Variants. Biol. Pharm. Bull. 2016, 39, 1748–1759. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Eguchi, S.; Ieiri, I. Impact of genetic polymorphisms in CYP2C9 and CYP2C19 on the pharmacokinetics of clinically used drugs. Drug Metab. Pharmacokinet. 2013, 28, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, S.R.; Muhammad, S.; Eidens, M.; Klemm, M.; Khan, D.; Efferth, T.; Weisshaar, M.P. Pharmacogenetic prediction of individual variability in drug response based on CYP2D6, CYP2C9 and CYP2C19 genetic polymorphisms. Curr. Drug Metab. 2014, 15, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.O.; Veronese, M.E.; Birkett, D.J. In Vitro Approaches for the Prediction of Human Drug Metabolism. Annu. Rep. Med. Chem. 1994, 29, 307–316. [Google Scholar]
- Miners, J.O.; Birkett, D.J. Use of tolbutamide as a substrate probe for human hepatic cytochrome P450 2C9. Methods Enzymol. 1996, 272, 139–145. [Google Scholar] [PubMed]
- Miners, J.O.; Coulter, S.; Birkett, D.J.; Goldstein, J.A. Torsemide metabolism by CYP2C9 variants and other human CYP2C subfamily enzymes. Pharmacogenetics 2000, 10, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.O.; Smith, K.J.; Robson, R.A.; McManus, M.E.; Veronese, M.E.; Birkett, D.J. Tolbutamide hydroxylation by human liver microsomes. Kinetic characterisation and relationship to other cytochrome P-450 dependent xenobiotic oxidations. Biochem. Pharmacol. 1988, 37, 1137–1144. [Google Scholar] [CrossRef]
- Sykes, M.J.; McKinnon, R.A.; Miners, J.O. Prediction of Metabolism by Cytochrome P450 2C9: Alignment and Docking Studies of a Validated Database of Substrates. J. Med. Chem. 2008, 51, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, J.; Huang, S.Q.; Su, H.H.; Zhou, S.F. Genetic polymorphism of the human cytochrome P450 2C9 gene and its clinical significance. Curr. Drug Metab. 2009, 10, 781–834. [Google Scholar] [CrossRef] [PubMed]
- Veronese, M.E.; Doecke, C.J.; Mackenzie, P.I.; McManus, M.E.; Miners, J.O.; Rees, D.L.; Gasser, R.; Meyer, U.A.; Birkett, D.J. Site-directed mutation studies of human liver cytochrome P-450 isoenzymes in the CYP2C subfamily. Biochem. J. 1993, 289, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Doecke, C.J.; Veronese, M.E.; Pond, S.M.; Miners, J.O.; Birkett, D.J.; Sansom, L.N.; McManus, M.E. Relationship between phenytoin and tolbutamide hydroxylations in human liver microsomes. Br. J. Clin. Pharmacol. 1991, 31, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.O.; Rees, D.L.; Valente, L.; Veronese, M.E.; Birkett, D.J. Human hepatic cytochrome P450 2C9 catalyzes the rate-limiting pathway of torsemide metabolism. J. Pharmacol. Exp. Ther. 1995, 272, 1076–1081. [Google Scholar] [PubMed]
- Yasar, U.; Tybring, G.; Hidestrand, M.; Oscarson, M.; Ingelman-Sundberg, M.; Dahl, M.L.; Eliasson, E. Role of CYP2C9 polymorphism in losartan oxidation. Drug Metab. Dispos. 2001, 29, 1051–1056. [Google Scholar] [PubMed]
- Lee, C.R.; Goldstein, J.A.; Pieper, J.A. Cytochrome P450 2C9 polymorphisms: A comprehensive review of the in vitro and human data. Pharmacogenetics 2002, 12, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.D.; Hamman, M.A.; Rettie, A.E.; Wienkers, L.C.; Trager, W.F.; Vandenbranden, M.; Wrighton, S.A. Relationships between the levels of cytochrome P4502C9 and its prototypic catalytic activities in human liver microsomes. Drug Metab. Dispos. 1994, 22, 975–978. [Google Scholar] [PubMed]
- Fuhr, U.; Jetter, A.; Kirchheiner, J. Appropriate Phenotyping Procedures for Drug Metabolizing Enzymes and Transporters in Humans and Their Simultaneous Use in the “Cocktail” Approach. Clin. Pharmacol. Ther. 2007, 81, 270–283. [Google Scholar] [CrossRef] [PubMed]
- De Andres, F.; Teran, S.; Bovera, M.; Farinas, H.; Teran, E.; A, L.L. Multiplex Phenotyping for Systems Medicine: A One-Point Optimized Practical Sampling Strategy for Simultaneous Estimation of CYP1A2, CYP2C9, CYP2C19, and CYP2D6 Activities Using a Cocktail Approach. Omics 2016, 20, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Nafziger, A.N.; Gaedigk, A.; Dickmann, L.J.; Rettie, A.E.; Bertino, J.S., Jr. Effects of oral vitamin K on S- and R-warfarin pharmacokinetics and pharmacodynamics: Enhanced safety of warfarin as a CYP2C9 probe. J. Clin. Pharmacol. 2001, 41, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Wester, M.R.; Yano, J.K.; Schoch, G.A.; Yang, C.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The structure of human cytochrome P450 2C9 complexed with flurbiprofen at 2.0-A resolution. J. Biol. Chem. 2004, 279, 35630–35637. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, O. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J. Biol. Chem. 1992, 267, 83–90. [Google Scholar] [PubMed]
- Nair, P.C.; McKinnon, R.A.; Miners, J.O. Cytochrome P450 structure-function: Insights from molecular dynamics simulations. Drug Metab. Rev. 2016, 48, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Ridderstrom, M.; Masimirembwa, C.; Trump-Kallmeyer, S.; Ahlefelt, M.; Otter, C.; Andersson, T.B. Arginines 97 and 108 in CYP2C9 are important determinants of the catalytic function. Biochem. Biophys. Res. Commun. 2000, 270, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Tai, G.; Dickmann, L.J.; Matovic, N.; DeVoss, J.J.; Gillam, E.M.; Rettie, A.E. Re-engineering of CYP2C9 to probe acid-base substrate selectivity. Drug Metab. Dispos. 2008, 36, 1992–1997. [Google Scholar] [CrossRef] [PubMed]
- Dickmann, L.J.; Locuson, C.W.; Jones, J.P.; Rettie, A.E. Differential roles of Arg97, Asp293, and Arg108 in enzyme stability and substrate specificity of CYP2C9. Mol. Pharmacol. 2004, 65, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Haining, R.L.; Jones, J.P.; Henne, K.R.; Fisher, M.B.; Koop, D.R.; Trager, W.F.; Rettie, A.E. Enzymatic Determinants of the Substrate Specificity of CYP2C9: Role of B’−C Loop Residues in Providing the π-Stacking Anchor Site for Warfarin Binding. Biochemistry 1999, 38, 3285–3292. [Google Scholar] [CrossRef] [PubMed]
- Melet, A.; Assrir, N.; Jean, P.; Pilar Lopez-Garcia, M.; Marques-Soares, C.; Jaouen, M.; Dansette, P.M.; Sari, M.A.; Mansuy, D. Substrate selectivity of human cytochrome P450 2C9: Importance of residues 476, 365, and 114 in recognition of diclofenac and sulfaphenazole and in mechanism-based inactivation by tienilic acid. Arch. Biochem. Biophys. 2003, 409, 80–91. [Google Scholar] [CrossRef]
- Flanagan, J.U.; McLaughlin, L.A.; Paine, M.J.; Sutcliffe, M.J.; Roberts, G.C.; Wolf, C.R. Role of conserved Asp293 of cytochrome P450 2C9 in substrate recognition and catalytic activity. Biochem. J. 2003, 370, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Cosme, J.; Ward, A.; Angove, H.C.; Matak Vinkovic, D.; Jhoti, H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 2003, 424, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, K.; Adachi, M.; Matsuzawa, Y.; Zhang, Q.; Kuroki, R.; Saito, Y.; Shah, M.B. Structural Basis of Single-Nucleotide Polymorphisms in Cytochrome P450 2C9. Biochemistry 2017, 56, 5460–5480. [Google Scholar] [CrossRef] [PubMed]
- Branden, G.; Sjogren, T.; Schnecke, V.; Xue, Y. Structure-based ligand design to overcome CYP inhibition in drug discovery projects. Drug Discov. Today 2014, 19, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.G.; Cheesman, M.J.; Primak, A.; Bowman, M.K.; Atkins, W.M.; Rettie, A.E. Intramolecular Heme Ligation of the Cytochrome P450 2C9 R108H Mutant Demonstrates Pronounced Conformational Flexibility of the B−C Loop Region: Implications for Substrate Binding. Biochemistry 2010, 49, 8700–8708. [Google Scholar] [CrossRef] [PubMed]
- Rettie, A.E.; Korzekwa, K.R.; Kunze, K.L.; Lawrence, R.F.; Eddy, A.C.; Aoyama, T.; Gelboin, H.V.; Gonzalez, F.J.; Trager, W.F. Hydroxylation of warfarin by human cDNA-expressed cytochrome P-450: A role for P-4502C9 in the etiology of (S)-warfarin-drug interactions. Chem. Res. Toxicol. 1992, 5, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Jonas, D.E.; McLeod, H.L. Genetic and clinical factors relating to warfarin dosing. Trends Pharmacol. Sci. 2009, 30, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, H.H.; Flinois, J.P.; Beaune, P.H. Cytochrome P4502C9 is the principal catalyst of racemic acenocoumarol hydroxylation reactions in human liver microsomes. Drug Metab. Dispos. 2000, 28, 1284–1290. [Google Scholar] [PubMed]
- Hermans, J.J.; Thijssen, H.H. Human liver microsomal metabolism of the enantiomers of warfarin and acenocoumarol: P450 isozyme diversity determines the differences in their pharmacokinetics. Br. J. Pharmacol. 1993, 110, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Ufer, M.; Svensson, J.O.; Krausz, K.W.; Gelboin, H.V.; Rane, A.; Tybring, G. Identification of cytochromes P450 2C9 and 3A4 as the major catalysts of phenprocoumon hydroxylation in vitro. Eur. J. Clin. Pharmacol. 2004, 60, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Dzeshka, M.S.; Lip, G.Y. Non-vitamin K oral anticoagulants in atrial fibrillation: Where are we now? Trends Cardiovasc. Med. 2015, 25, 315–336. [Google Scholar] [CrossRef] [PubMed]
- Tornio, A.; Niemi, M.; Neuvonen, P.J.; Backman, J.T. Drug interactions with oral antidiabetic agents: Pharmacokinetic mechanisms and clinical implications. Trends Pharmacol. Sci. 2012, 33, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Schopman, J.E.; Simon, A.C.; Hoefnagel, S.J.; Hoekstra, J.B.; Scholten, R.J.; Holleman, F. The incidence of mild and severe hypoglycaemia in patients with type 2 diabetes mellitus treated with sulfonylureas: A systematic review and meta-analysis. Diabetes Metab. Res. Rev. 2014, 30, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Elliot, D.J.; Suharjono; Lewis, B.C.; Gillam, E.M.J.; Birkett, D.J.; Gross, A.S.; Miners, J.O. Identification of the human cytochromes P450 catalysing the rate-limiting pathways of gliclazide elimination. Br. J. Clin. Pharmacol. 2007, 64, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Zhang, Y.F.; Chen, X.Y.; Zhao, X.H.; Li, G.X.; Zhong, D.F. The effects of CYP2C9 and CYP2C19 genetic polymorphisms on the pharmacokinetics and pharmacodynamics of glipizide in Chinese subjects. Eur. J. Clin. Pharmacol. 2010, 66, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Manolopoulos, V.G.; Ragia, G.; Tavridou, A. Pharmacogenomics of oral antidiabetic medications: Current data and pharmacoepigenomic perspective. Pharmacogenomics 2011, 12, 1161–1191. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.H. Cytochrome P450 isozymes and antiepileptic drug interactions. Epilepsia 1995, 36 (Suppl. 5), S8–S13. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.I.; Kim, M.J.; Chung, E.K.; Lee, H.I.; Jang, C.G.; Bae, J.W.; Lee, S.Y. CYP2C9 3 and 13 alleles significantly affect the pharmacokinetics of irbesartan in healthy Korean subjects. Eur. J. Clin. Pharmacol. 2012, 68, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Uchida, S.; Watanabe, H.; Nishio, S.; Hashimoto, H.; Yamazaki, K.; Hayashi, H.; Ohashi, K. Altered pharmacokinetics and excessive hypotensive effect of candesartan in a patient with the CYP2C91/3 genotype. Clin. Pharmacol. Ther. 2003, 74, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.; Kawashita, H.; Masuda, N.; Saxer, C.; Niina, M.; Nagae, Y.; Iwasaki, K. Identification of cytochrome P450 forms involved in the 4-hydroxylation of valsartan, a potent and specific angiotensin II receptor antagonist, in human liver microsomes. Xenobiotica 2005, 35, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Luo, Z.; Liu, Y.; Sun, M.; Zheng, L.; Chen, Y.; Li, Y.; Wang, H.; Chen, L.; Wu, M.; et al. Drug Interactions with Angiotensin Receptor Blockers: Role of Human Cytochromes P450. Curr. Drug Metab. 2016, 17, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Shahabi, P.; Siest, G.; Meyer, U.A.; Visvikis-Siest, S. Human cytochrome P450 epoxygenases: Variability in expression and role in inflammation-related disorders. Pharmacol. Ther. 2014, 144, 134–161. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, R.A.; Trager, W.F.; Rettie, A.E.; Goulart, D.A. Interaction of amiodarone with racemic warfarin and its separated enantiomorphs in humans. Clin. Pharmacol. Ther. 1987, 42, 290–294. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.G.; Au, N.T.; Wittkowsky, A.K.; Rettie, A.E. Warfarin-amiodarone drug-drug interactions: Determination of [I]u/KI,u for amiodarone and its plasma metabolites. Clin. Pharmacol. Ther. 2012, 91, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Zilly, W.; Breimer, D.D.; Richter, E. Induction of drug metabolism in man after rifampicin treatment measured by increased hexobarbital and tolbutamide clearance. Eur. J. Clin. Pharmacol. 1975, 9, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Heimark, L.D.; Gibaldi, M.; Trager, W.F.; O’Reilly, R.A.; Goulart, D.A. The mechanism of the warfarin-rifampin drug interaction in humans. Clin. Pharmacol. Ther. 1987, 42, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Bertilsson, G.; Heidrich, J.; Svensson, K.; Asman, M.; Jendeberg, L.; Sydow-Backman, M.; Ohlsson, R.; Postlind, H.; Blomquist, P.; Berkenstam, A. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc. Natl. Acad. Sci. USA 1998, 95, 12208–12213. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.M.; McKee, D.D.; Watson, M.A.; Willson, T.M.; Moore, J.T.; Kliewer, S.A. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Investig. 1998, 102, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ferguson, S.S.; Negishi, M.; Goldstein, J.A. Induction of human CYP2C9 by rifampicin, hyperforin, and phenobarbital is mediated by the pregnane X receptor. J. Pharmacol. Exp. Ther. 2004, 308, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.S.; LeCluyse, E.L.; Negishi, M.; Goldstein, J.A. Regulation of human CYP2C9 by the constitutive androstane receptor: Discovery of a new distal binding site. Mol. Pharmacol. 2002, 62, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Gerbal-Chaloin, S.; Daujat, M.; Pascussi, J.M.; Pichard-Garcia, L.; Vilarem, M.J.; Maurel, P. Transcriptional regulation of CYP2C9 gene. Role of glucocorticoid receptor and constitutive androstane receptor. J. Biol. Chem. 2002, 277, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Sahi, J.; Shord, S.S.; Lindley, C.; Ferguson, S.; LeCluyse, E.L. Regulation of cytochrome P450 2C9 expression in primary cultures of human hepatocytes. J. Biochem. Mol. Toxicol. 2009, 23, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Ranade, A.; Venkataramanan, R.; Strom, S.; Chupka, J.; Ekins, S.; Schuetz, E.; Bachmann, K. A comprehensive in vitro and in silico analysis of antibiotics that activate pregnane X receptor and induce CYP3A4 in liver and intestine. Drug Metab. Dispos. 2008, 36, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.J.; Sakamuru, S.; Huang, R.; Moeller, T.A.; Shinn, P.; Vanleer, D.; Auld, D.S.; Austin, C.P.; Xia, M. Identification of clinically used drugs that activate pregnane X receptors. Drug Metab. Dispos. 2011, 39, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Stage, T.; Wong, S.; Kroetz, D.; Khojasteh, C. Dicloxacillin and flucloxacillin induce CYP3A4 and CYP2C9 in human hepatocytes. Clin. Pharmacol. Ther. 2017, 101, S47–S48. [Google Scholar]
- Pottegard, A.; Henriksen, D.P.; Madsen, K.G.; Hellfritzsch, M.; Damkier, P.; Stage, T.B. Change in International Normalized Ratio among Patients Treated with Dicloxacillin and Vitamin K Antagonists. JAMA 2015, 314, 296–297. [Google Scholar] [CrossRef] [PubMed]
- Merwick, A.; Hannon, N.; Kelly, P.J.; O’Rourke, K. Warfarin-flucloxacillin interaction presenting as cardioembolic ischemic stroke. Eur. J. Clin. Pharmacol. 2010, 66, 643–644. [Google Scholar] [CrossRef] [PubMed]
- Mwinyi, J.; Cavaco, I.; Yurdakok, B.; Mkrtchian, S.; Ingelman-Sundberg, M. The ligands of estrogen receptor α regulate cytochrome P4502C9 (CYP2C9) expression. J. Pharmacol. Exp. Ther. 2011, 338, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Johansson, I.; Christensen, M.; Rane, A.; Eliasson, E. The impact of CYP2C9 genetics and oral contraceptives on cytochrome P450 2C9 phenotype. Drug Metab. Dispos. 2004, 32, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.; Poffenbarger, P.L. Pharmacogenetics of tolbutamide metabolism in humans. Diabetes 1979, 28, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Pastewka, J.; Gelboin, H.V.; Gonzalez, F.J. cDNA and Amino-Acid-Sequences of 2 Members of the Human P450IIC Gene Subfamily. Nucleic Acids Res. 1987, 15, 10053–10054. [Google Scholar] [CrossRef] [PubMed]
- Meehan, R.R.; Gosden, J.R.; Rout, D.; Hastie, N.D.; Friedberg, T.; Adesnik, M.; Buckland, R.; van Heyningen, V.; Fletcher, J.; Spurr, N.K.; et al. Human cytochrome P450 PB-1: A multigene family involved in mephenytoin and steroid oxidations that maps to chromosome 10. Am. J. Hum. Genet. 1988, 42, 26–37. [Google Scholar] [PubMed]
- Rettie, A.E.; Wienkers, L.C.; Gonzalez, F.J.; Trager, W.F.; Korzekwa, K.R. Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics 1994, 4, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Sullivan-Klose, T.H.; Ghanayem, B.I.; Bell, D.A.; Zhang, Z.Y.; Kaminsky, L.S.; Shenfield, G.M.; Miners, J.O.; Birkett, D.J.; Goldstein, J.A. The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics 1996, 6, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Haining, R.L.; Hunter, A.P.; Veronese, M.E.; Trager, W.F.; Rettie, A.E. Allelic variants of human cytochrome P450 2C9: Baculovirus-mediated expression, purification, structural characterization, substrate stereoselectivity, and prochiral selectivity of the wild-type and I359L mutant forms. Arch. Biochem. Biophys. 1996, 333, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Furuya, H.; FernandezSalguero, P.; Gregory, W.; Taber, H.; Steward, A.; Gonzalez, F.J.; Idle, J.R. Genetic polymorphism of CYP2C9 and its effect on warfarin maintenance dose requirement in patients undergoing anticoagulation therapy. Pharmacogenetics 1995, 5, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Steward, D.J.; Haining, R.L.; Henne, K.R.; Davis, G.; Rushmore, T.H.; Trager, W.F.; Rettie, A.E. Genetic association between sensitivity to warfarin and expression of CYP2C9*3. Pharmacogenetics 1997, 7, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.P.; Day, C.P.; Kesteven, P.J.L.; Daly, A.K. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet 1999, 353, 717–719. [Google Scholar] [CrossRef]
- Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Worldwide Distribution of Cytochrome P450 Alleles: A Meta-analysis of Population-scale Sequencing Projects. Clin. Pharmacol. Ther. 2017, 102, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Allabi, A.C.; Gala, J.L.; Horsmans, Y.; Babaoglu, M.O.; Bozkurt, A.; Heusterspreute, M.; Yasar, U. Functional impact of CYP2C9*5, CYP2C9*6, CYP2C9*8, and CYP2C9*11 in vivo among black Africans. Clin. Pharmacol. Ther. 2004, 76, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Ieiri, I.; Wilkinson, G.R.; Mayo, G.; Kashima, T.; Kimura, S.; Otsubo, K.; Echizen, H. 5′-Flanking region polymorphisms of CYP2C9 and their relationship to S-warfarin metabolism in white and Japanese patients. Blood 2004, 103, 3055–3057. [Google Scholar] [CrossRef] [PubMed]
- Tai, G.; Farin, F.; Rieder, M.J.; Dreisbach, A.W.; Veenstra, D.L.; Verlinde, C.L.; Rettie, A.E. In vitro and in vivo effects of the CYP2C9*11 polymorphism on warfarin metabolism and dose. Pharmacogenet. Genom. 2005, 15, 475–481. [Google Scholar] [CrossRef]
- Hernandez, W.; Gamazon, E.R.; Aquino-Michaels, K.; Patel, S.; O’Brien, T.J.; Harralson, A.F.; Kittles, R.A.; Barbour, A.; Tuck, M.; McIntosh, S.D.; et al. Ethnicity-specific pharmacogenetics: The case of warfarin in African Americans. Pharmacogenom. J. 2014, 14, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.P.; Xu, R.A.; Hu, L.M.; Wang, S.H.; Geng, P.W.; Yang, J.F.; Yang, L.P.; Qian, J.C.; Wang, Z.S.; Zhu, G.H.; et al. CYP2C9 polymorphism analysis in Han Chinese populations: Building the largest allele frequency database. Pharmacogenom. J. 2014, 14, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Allabi, A.C.; Gala, J.L.; Horsmans, Y. CYP2C9, CYP2C19, ABCB1 (MDR1) genetic polymorphisms and phenytoin metabolism in a Black Beninese population. Pharmacogenet. Genom. 2005, 15, 779–786. [Google Scholar] [CrossRef]
- Blaisdell, J.; Jorge-Nebert, L.F.; Coulter, S.; Ferguson, S.S.; Lee, S.J.; Chanas, B.; Xi, T.; Mohrenweiser, H.; Ghanayem, B.; Goldstein, J.A. Discovery of new potentially defective alleles of human CYP2C9. Pharmacogenetics 2004, 14, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jeong, H.; Takahashi, H.; Drozda, K.; Patel, S.R.; Shapiro, N.L.; Nutescu, E.A.; Cavallari, L.H. Decreased warfarin clearance associated with the CYP2C9 R150H (*8) polymorphism. Clin. Pharmacol. Ther. 2012, 91, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Niinuma, Y.; Saito, T.; Takahashi, M.; Tsukada, C.; Ito, M.; Hirasawa, N.; Hiratsuka, M. Functional characterization of 32 CYP2C9 allelic variants. Pharmacogenom. J. 2014, 14, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Dickmann, L.J.; Rettie, A.E.; Kneller, M.B.; Kim, R.B.; Wood, A.J.; Stein, C.M.; Wilkinson, G.R.; Schwarz, U.I. Identification and functional characterization of a new CYP2C9 variant (CYP2C9*5) expressed among African Americans. Mol. Pharmacol. 2001, 60, 382–387. [Google Scholar] [PubMed]
- Kidd, R.S.; Curry, T.B.; Gallagher, S.; Edeki, T.; Blaisdell, J.; Goldstein, J.A. Identification of a null allele of CYP2C9 in an African- American exhibiting toxicity to phenytoin. Pharmacogenetics 2001, 11, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Reynald, R.L.; Sansen, S.; Stout, C.D.; Johnson, E.F. Structural characterization of human cytochrome P450 2C19: Active site differences between P450s 2C8, 2C9, and 2C19. J. Biol. Chem. 2012, 287, 44581–44591. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.P.; Wang, Y.H.; Wang, S.H.; Geng, P.W.; Hu, L.M.; Hu, G.X.; Cai, J.P. In vitro functional characterization of 37 CYP2C9 allelic isoforms found in Chinese Han population. Acta Pharmacol. Sin. 2013, 34, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Fohner, A.E.; Robinson, R.; Yracheta, J.; Dillard, D.A.; Schilling, B.; Khan, B.; Hopkins, S.; Boyer, B.; Black, J.; Wiener, H.; et al. Variation in genes controlling warfarin disposition and response in American Indian and Alaska Native people: CYP2C9, VKORC1, CYP4F2, CYP4F11, GGCX. Pharmacogenet. Genom. 2015, 25, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Rosdi, R.A.; Mohd Yusoff, N.; Ismail, R.; Soo Choon, T.; Saleem, M.; Musa, N.; Yusoff, S. High allele frequency of CYP2C9*3 (rs1057910) in a Negrito’s subtribe population in Malaysia; Aboriginal people of Jahai. Ann. Hum. Biol. 2016, 43, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Macias, M.; Lazalde-Ramos, B.P.; Galaviz-Hernandez, C.; Rangel-Villalobos, H.; Salazar-Flores, J.; Martinez-Sevilla, V.M.; Martinez-Fierro, M.L.; Dorado, P.; Wong, M.L.; Licinio, J.; et al. Influence of admixture components on CYP2C9*2 allele frequency in eight indigenous populations from Northwest Mexico. Pharmacogenom. J. 2013, 13, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Henderson, L.M.; Claw, K.G.; Woodahl, E.L.; Robinson, R.F.; Boyer, B.B.; Burke, W.; Thummel, K.E. P450 Pharmacogenetics in Indigenous North American Populations. J. Personal. Med. 2018. submitted for publication. [Google Scholar]
- Cespedes-Garro, C.; Fricke-Galindo, I.; Naranjo, M.E.; Rodrigues-Soares, F.; Farinas, H.; de Andres, F.; Lopez-Lopez, M.; Penas-Lledo, E.M.; Llerena, A. Worldwide interethnic variability and geographical distribution of CYP2C9 genotypes and phenotypes. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Thusberg, J.; Olatubosun, A.; Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 2011, 32, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Jarrar, Y.B.; Lee, S.J. Molecular functionality of CYP2C9 polymorphisms and their influence on drug therapy. Drug Metabol. Drug Interact. 2014, 29, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Elliot, D.J.; Knights, K.M.; Mackenzie, P.I.; Miners, J.O. The “albumin effect” and in vitro-in vivo extrapolation: Sequestration of long-chain unsaturated fatty acids enhances phenytoin hydroxylation by human liver microsomal and recombinant cytochrome P450 2C9. Drug Metab. Dispos. 2008, 36, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Crespi, C.L.; Miller, V.P. The R144C change in the CYP2C9*2 allele alters interaction of the cytochrome P450 with NADPH:cytochrome P450 oxidoreductase. Pharmacogenetics 1997, 7, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Locuson, C.W.; Tracy, T.S. Polymorphic Variants of CYP2C9: Mechanisms Involved in Reduced Catalytic Activity. Mol. Pharmacol. 2007, 72, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Sano, E.; Li, W.; Yuki, H.; Liu, X.; Furihata, T.; Kobayashi, K.; Chiba, K.; Neya, S.; Hoshino, T. Mechanism of the decrease in catalytic activity of human cytochrome P450 2C9 polymorphic variants investigated by computational analysis. J. Comput. Chem. 2010, 31, 2746–2758. [Google Scholar] [CrossRef] [PubMed]
- Lertkiatmongkol, P.; Assawamakin, A.; White, G.; Chopra, G.; Rongnoparut, P.; Samudrala, R.; Tongsima, S. Distal effect of amino acid substitutions in CYP2C9 polymorphic variants causes differences in interatomic interactions against (S)-warfarin. PLoS ONE 2013, 8, e74053. [Google Scholar] [CrossRef]
- Banu, H.; Renuka, N.; Vasanthakumar, G. Reduced catalytic activity of human CYP2C9 natural alleles for gliclazide: Molecular dynamics simulation and docking studies. Biochimie 2011, 93, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- DeLozier, T.C.; Lee, S.C.; Coulter, S.J.; Goh, B.C.; Goldstein, J.A. Functional characterization of novel allelic variants of CYP2C9 recently discovered in southeast Asians. J. Pharmacol. Exp. Ther. 2005, 315, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, D.L.; Blough, D.K.; Higashi, M.K.; Farin, F.M.; Srinouanprachan, S.; Rieder, M.J.; Rettie, A.E. CYP2C9 haplotype structure in European American warfarin patients and association with clinical outcomes. Clin. Pharmacol. Ther. 2005, 77, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.A.; Rettie, A.E.; Rieder, M.J.; Cabacungan, E.T.; Hines, R.N. Novel CYP2C9 promoter variants and assessment of their impact on gene expression. Mol. Pharmacol. 2008, 73, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.S.; Urban, T.J.; Lamba, J.K.; Birnbaum, A.K.; Remmel, R.P.; Subramanian, M.; Strom, S.; You, J.H.; Kasperaviciute, D.; Catarino, C.B.; et al. CYP2C9*1B promoter polymorphisms, in linkage with CYP2C19*2, affect phenytoin autoinduction of clearance and maintenance dose. J. Pharmacol. Exp. Ther. 2010, 332, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Sun, X.; Gong, Y.; Gawronski, B.E.; Langaee, T.Y.; Shahin, M.H.; Khalifa, S.I.; Johnson, J.A. CYP2C9 promoter variable number tandem repeat polymorphism regulates mRNA expression in human livers. Drug Metab. Dispos. 2012, 40, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Shintani, M.; Ieiri, I.; Inoue, K.; Mamiya, K.; Ninomiya, H.; Tashiro, N.; Higuchi, S.; Otsubo, K. Genetic polymorphisms and functional characterization of the 5′-flanking region of the human CYP2C9 gene: In vitro and in vivo studies. Clin. Pharmacol. Ther. 2001, 70, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Cavallari, L.H.; Vaynshteyn, D.; Freeman, K.M.; Wang, D.; Perera, M.A.; Takahashi, H.; Drozda, K.; Patel, S.R.; Jeong, H. CYP2C9 promoter region single-nucleotide polymorphisms linked to the R150H polymorphism are functional suggesting their role in CYP2C9*8-mediated effects. Pharmacogenet. Genom. 2013, 23, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.A.; Gamazon, E.; Cavallari, L.H.; Patel, S.R.; Poindexter, S.; Kittles, R.A.; Nicolae, D.; Cox, N.J. The missing association: Sequencing-based discovery of novel SNPs in VKORC1 and CYP2C9 that affect warfarin dose in African Americans. Clin. Pharmacol. Ther. 2011, 89, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Yasar, U.; Lundgren, S.; Eliasson, E.; Bennet, A.; Wiman, B.; de Faire, U.; Rane, A. Linkage between the CYP2C8 and CYP2C9 genetic polymorphisms. Biochem. Biophys. Res. Commun. 2002, 299, 25–28. [Google Scholar] [CrossRef]
- Speed, W.C.; Kang, S.P.; Tuck, D.P.; Harris, L.N.; Kidd, K.K. Global variation in CYP2C8-CYP2C9 functional haplotypes. Pharmacogenom. J. 2009, 9, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, K.; Fukushima-Uesaka, H.; Tohkin, M.; Hasegawa, R.; Kajio, H.; Kuzuya, N.; Yasuda, K.; Kawamoto, M.; Kamatani, N.; Suzuki, K.; et al. Four novel defective alleles and comprehensive haplotype analysis of CYP2C9 in Japanese. Pharmacogenet. Genom. 2006, 16, 497–514. [Google Scholar] [CrossRef] [PubMed]
- Gage, B.F.; Eby, C.; Milligan, P.E.; Banet, G.A.; Duncan, J.R.; McLeod, H.L. Use of pharmacogenetics and clinical factors to predict the maintenance dose of warfarin. Thromb. Haemost. 2004, 91, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.; Halsall, D.; Baglin, T. Influence of cytochrome P-450CYP2C9 polymorphisms on warfarin sensitivity and risk of over-anticoagulation in patients on long-term treatment. Blood 2000, 96, 1816–1819. [Google Scholar] [PubMed]
- Higashi, M.K.; Veenstra, D.L.; Kondo, L.M.L.; Wittkowsky, A.K.; Srinouanprachanh, S.L.; Farin, F.M.; Rettie, A.E. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002, 287, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Wadelius, M.; Sorlin, K.; Wallerman, O.; Karlsson, J.; Yue, Q.Y.; Magnusson, P.K.; Wadelius, C.; Melhus, H. Warfarin sensitivity related to CYP2C9, CYP3A5, ABCB1 (MDR1) and other factors. Pharmacogenom. J. 2004, 4, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Parra, E.J.; Botton, M.R.; Perini, J.A.; Krithika, S.; Bourgeois, S.; Johnson, T.A.; Tsunoda, T.; Pirmohamed, M.; Wadelius, M.; Limdi, N.A.; et al. Genome-wide association study of warfarin maintenance dose in a Brazilian sample. Pharmacogenomics 2015, 16, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, N.; Wallentin, L.; Berglund, L.; Axelsson, T.; Connolly, S.; Eikelboom, J.; Ezekowitz, M.; Oldgren, J.; Pare, G.; Reilly, P.; et al. Genetic determinants of warfarin maintenance dose and time in therapeutic treatment range: A RE-LY genomics substudy. Pharmacogenomics 2016, 17, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M.; Johnson, J.A.; Langaee, T.Y.; Feng, H.; Stanaway, I.B.; Schwarz, U.I.; Ritchie, M.D.; Stein, C.M.; Roden, D.M.; Smith, J.D.; et al. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood 2008, 112, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, F.; McGinnis, R.; Bourgeois, S.; Barnes, C.; Eriksson, N.; Soranzo, N.; Whittaker, P.; Ranganath, V.; Kumanduri, V.; McLaren, W.; et al. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet. 2009, 5, e1000433. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, S.; Jorgensen, A.; Zhang, E.J.; Hanson, A.; Gillman, M.S.; Bumpstead, S.; Toh, C.H.; Williamson, P.; Daly, A.K.; Kamali, F.; et al. A multi-factorial analysis of response to warfarin in a UK prospective cohort. Genome Med. 2016, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, A.L.; FitzGerald, R.J.; Oyee, J.; Pirmohamed, M.; Williamson, P.R. Influence of CYP2C9 and VKORC1 on patient response to warfarin: A systematic review and meta-analysis. PLoS ONE 2012, 7, e44064. [Google Scholar] [CrossRef] [PubMed]
- Ufer, M. Comparative pharmacokinetics of vitamin K antagonists: Warfarin, phenprocoumon and acenocoumarol. Clin. Pharmacokinet. 2005, 44, 1227–1246. [Google Scholar] [CrossRef] [PubMed]
- Van Schie, R.M.; Wessels, J.A.; le Cessie, S.; de Boer, A.; Schalekamp, T.; van der Meer, F.J.; Verhoef, T.I.; van Meegen, E.; Rosendaal, F.R.; Maitland-van der Zee, A.H. Loading and maintenance dose algorithms for phenprocoumon and acenocoumarol using patient characteristics and pharmacogenetic data. Eur. Heart J. 2011, 32, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, T.I.; Ragia, G.; de Boer, A.; Barallon, R.; Kolovou, G.; Kolovou, V.; Konstantinides, S.; Le Cessie, S.; Maltezos, E.; van der Meer, F.J.; et al. A randomized trial of genotype-guided dosing of acenocoumarol and phenprocoumon. N. Engl. J. Med. 2013, 369, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.L.; Visser, L.E.; Trienekens, P.H.; Hofman, A.; van Schaik, R.H.; Stricker, B.H. Cytochrome P450 2C9 *2 and *3 polymorphisms and the dose and effect of sulfonylurea in type II diabetes mellitus. Clin. Pharmacol. Ther. 2008, 83, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Donnelly, L.; Burch, L.; Tavendale, R.; Doney, A.S.; Leese, G.; Hattersley, A.T.; McCarthy, M.I.; Morris, A.D.; Lang, C.C.; et al. Loss-of-function CYP2C9 variants improve therapeutic response to sulfonylureas in type 2 diabetes: A Go-DARTS study. Clin. Pharmacol. Ther. 2010, 87, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Holstein, A.; Plaschke, A.; Ptak, M.; Egberts, E.H.; El-Din, J.; Brockmoller, J.; Kirchheiner, J. Association between CYP2C9 slow metabolizer genotypes and severe hypoglycaemia on medication with sulphonylurea hypoglycaemic agents. Br. J. Clin. Pharmacol. 2005, 60, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Ragia, G.; Petridis, I.; Tavridou, A.; Christakidis, D.; Manolopoulos, V.G. Presence of CYP2C9*3 allele increases risk for hypoglycemia in Type 2 diabetic patients treated with sulfonylureas. Pharmacogenomics 2009, 10, 1781–1787. [Google Scholar] [CrossRef] [PubMed]
- Gokalp, O.; Gunes, A.; Cam, H.; Cure, E.; Aydin, O.; Tamer, M.N.; Scordo, M.G.; Dahl, M.L. Mild hypoglycaemic attacks induced by sulphonylureas related to CYP2C9, CYP2C19 and CYP2C8 polymorphisms in routine clinical setting. Eur. J. Clin. Pharmacol. 2011, 67, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.T.; Zauber, A.G.; Hsu, M.; Breazna, A.; Hunter, D.J.; Rosenstein, R.B.; Eagle, C.J.; Hawk, E.T.; Bertagnolli, M.M. Cytochrome P450 2C9 variants influence response to celecoxib for prevention of colorectal adenoma. Gastroenterology 2009, 136, 2127–2136. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.D. Impact of CYP2C9 genotype on pharmacokinetics: Are all cyclooxygenase inhibitors the same? Drug Metab. Dispos. 2005, 33, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.; Blanco, G.; Ladero, J.M.; Garcia-Martin, E.; Taxonera, C.; Gamito, F.G.; Diaz-Rubio, M.; Agundez, J.A. Genetic predisposition to acute gastrointestinal bleeding after NSAIDs use. Br. J. Pharmacol. 2004, 141, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Pilotto, A.; Seripa, D.; Franceschi, M.; Scarcelli, C.; Colaizzo, D.; Grandone, E.; Niro, V.; Andriulli, A.; Leandro, G.; Di Mario, F.; et al. Genetic susceptibility to nonsteroidal anti-inflammatory drug-related gastroduodenal bleeding: Role of cytochrome P450 2C9 polymorphisms. Gastroenterology 2007, 133, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Figueiras, A.; Estany-Gestal, A.; Aguirre, C.; Ruiz, B.; Vidal, X.; Carvajal, A.; Salado, I.; Salgado-Barreira, A.; Rodella, L.; Moretti, U.; et al. CYP2C9 variants as a risk modifier of NSAID-related gastrointestinal bleeding: A case-control study. Pharmacogenet. Genom. 2016, 26, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.P.; Day, C.P.; Leathart, J.B.; Daly, A.K. Relationship of polymorphism in CYP2C9 to genetic susceptibility to diclofenac-induced hepatitis. Pharmacogenetics 2000, 10, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Urban, T.J.; Shen, Y.; Stolz, A.; Chalasani, N.; Fontana, R.J.; Rochon, J.; Ge, D.; Shianna, K.V.; Daly, A.K.; Lucena, M.I.; et al. Limited contribution of common genetic variants to risk for liver injury due to a variety of drugs. Pharmacogenet. Genom. 2012, 22, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Aynacioglu, A.S.; Brockmoller, J.; Bauer, S.; Sachse, C.; Guzelbey, P.; Ongen, Z.; Nacak, M.; Roots, I. Frequency of cytochrome P450 CYP2C9 variants in a Turkish population and functional relevance for phenytoin. Br. J. Clin. Pharmacol. 1999, 48, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Franco, V.; Perucca, E. CYP2C9 polymorphisms and phenytoin metabolism: Implications for adverse effects. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Dorado, P.; Lopez-Torres, E.; Penas-Lledo, E.M.; Martinez-Anton, J.; Llerena, A. Neurological toxicity after phenytoin infusion in a pediatric patient with epilepsy: Influence of CYP2C9, CYP2C19 and ABCB1 genetic polymorphisms. Pharmacogenom. J. 2013, 13, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.H.; Chang, W.C.; Lee, Y.S.; Wu, Y.Y.; Yang, C.H.; Ho, H.C.; Chen, M.J.; Lin, J.Y.; Hui, R.C.; Ho, J.C.; et al. Genetic variants associated with phenytoin-related severe cutaneous adverse reactions. JAMA 2014, 312, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.K.; Mahoney, H.C.; Levine, R.A. Phenytoin-induced chronic hepatitis. Dig. Dis. Sci. 1993, 38, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Yasar, U.; Forslund-Bergengren, C.; Tybring, G.; Dorado, P.; Llerena, A.; Sjoqvist, F.; Eliasson, E.; Dahl, M.L. Pharmacokinetics of losartan and its metabolite E-3174 in relation to the CYP2C9 genotype. Clin. Pharmacol. Ther. 2002, 71, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Sekino, K.; Kubota, T.; Okada, Y.; Yamada, Y.; Yamamoto, K.; Horiuchi, R.; Kimura, K.; Iga, T. Effect of the single CYP2C9*3 allele on pharmacokinetics and pharmacodynamics of losartan in healthy Japanese subjects. Eur. J. Clin. Pharmacol. 2003, 59, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, P.; Karlsson, J.; Kurland, L.; Lind, L.; Kahan, T.; Malmqvist, K.; Ohman, K.P.; Nystrom, F.; Melhus, H. The CYP2C9 genotype predicts the blood pressure response to irbesartan: Results from the Swedish Irbesartan Left Ventricular Hypertrophy Investigation vs Atenolol (SILVHIA) trial. J. Hypertens 2002, 20, 2089–2093. [Google Scholar] [CrossRef] [PubMed]
- Dingemanse, J.; van Giersbergen, P.L. Clinical pharmacology of bosentan, a dual endothelin receptor antagonist. Clin. Pharmacokinet. 2004, 43, 1089–1115. [Google Scholar] [CrossRef] [PubMed]
- Segal, E.S.; Valette, C.; Oster, L.; Bouley, L.; Edfjall, C.; Herrmann, P.; Raineri, M.; Kempff, M.; Beacham, S.; van Lierop, C. Risk management strategies in the postmarketing period: Safety experience with the US and European bosentan surveillance programmes. Drug Saf. 2005, 28, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Markova, S.M.; De Marco, T.; Bendjilali, N.; Kobashigawa, E.A.; Mefford, J.; Sodhi, J.; Le, H.; Zhang, C.; Halladay, J.; Rettie, A.E.; et al. Association of CYP2C9*2 with bosentan-induced liver injury. Clin. Pharmacol. Ther. 2013, 94, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Seyfarth, H.J.; Favreau, N.; Tennert, C.; Ruffert, C.; Halank, M.; Wirtz, H.; Mossner, J.; Rosendahl, J.; Kovacs, P.; Wittenburg, H. Genetic susceptibility to hepatoxicity due to bosentan treatment in pulmonary hypertension. Ann. Hepatol. 2014, 13, 803–809. [Google Scholar] [PubMed]
- Fleming, I. The pharmacology of the cytochrome P450 epoxygenase/soluble epoxide hydrolase axis in the vasculature and cardiovascular disease. Pharmacol. Rev. 2014, 66, 1106–1140. [Google Scholar] [CrossRef] [PubMed]
- Berlin, D.S.; Sangkuhl, K.; Klein, T.E.; Altman, R.B. PharmGKB summary: Cytochrome P450, family 2, subfamily J, polypeptide 2: CYP2J2. Pharmacogenet. Genom. 2011, 21, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Kamali, F.; Khan, T.I.; King, B.P.; Frearson, R.; Kesteven, P.; Wood, P.; Daly, A.K.; Wynne, H. Contribution of age, body size, and CYP2C9 genotype to anticoagulant response to warfarin. Clin. Pharmacol. Ther. 2004, 75, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Hillman, M.A.; Wilke, R.A.; Caldwell, M.D.; Berg, R.L.; Glurich, I.; Burmester, J.K. Relative impact of covariates in prescribing warfarin according to CYP2C9 genotype. Pharmacogenetics 2004, 14, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Sconce, E.A.; Khan, T.I.; Wynne, H.A.; Avery, P.; Monkhouse, L.; King, B.P.; Wood, P.; Kesteven, P.; Daly, A.K.; Kamali, F. The impact of CYP2C9 and VKORC1 genetic polymorphism and patient characteristics upon warfarin dose requirements: Proposal for a new dosing regimen. Blood 2005, 106, 2329–2333. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.; Peternel, P.; Stegnar, M.; Breskvar, K.; Dolzan, V. The influence of sequence variations in factor VII, gamma-glutamyl carboxylase and vitamin K epoxide reductase complex genes on warfarin dose requirement. Thromb. Haemost. 2006, 95, 782–787. [Google Scholar] [PubMed]
- Tham, L.S.; Goh, B.C.; Nafziger, A.; Guo, J.Y.; Wang, L.Z.; Soong, R.; Lee, S.C. A warfarin-dosing model in Asians that uses single-nucleotide polymorphisms in vitamin K epoxide reductase complex and cytochrome P450 2C9. Clin. Pharmacol. Ther. 2006, 80, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, M.D.; Berg, R.L.; Zhang, K.Q.; Glurich, I.; Schmelzer, J.R.; Yale, S.H.; Vidaillet, H.J.; Burmester, J.K. Evaluation of genetic factors for warfarin dose prediction. Clin. Med. Res. 2007, 5, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Horne, B.D.; Stevens, S.M.; Grove, A.S.; Barton, S.; Nicholas, Z.P.; Kahn, S.F.; May, H.T.; Samuelson, K.M.; Muhlestein, J.B.; et al. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation. Circulation 2007, 116, 2563–2570. [Google Scholar] [CrossRef] [PubMed]
- Millican, E.A.; Lenzini, P.A.; Milligan, P.E.; Grosso, L.; Eby, C.; Deych, E.; Grice, G.; Clohisy, J.C.; Barrack, R.L.; Burnett, R.S.; et al. Genetic-based dosing in orthopedic patients beginning warfarin therapy. Blood 2007, 110, 1511–1515. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shennan, M.; Reynolds, K.K.; Johnson, N.A.; Herrnberger, M.R.; Valdes, R., Jr.; Linder, M.W. Estimation of warfarin maintenance dose based on VKORC1 (−1639 G>A) and CYP2C9 genotypes. Clin. Chem. 2007, 53, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.H.; Wang, P.; Smith, A.; Haller, C.; Drake, K.; Linder, M.; Valdes, R., Jr. Dosing algorithm for warfarin using CYP2C9 and VKORC1 genotyping from a multi-ethnic population: Comparison with other equations. Pharmacogenomics 2008, 9, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Gage, B.F.; Eby, C.; Johnson, J.A.; Deych, E.; Rieder, M.J.; Ridker, P.M.; Milligan, P.E.; Grice, G.; Lenzini, P.; Rettie, A.E.; et al. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin. Pharmacol. Ther. 2008, 84, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.E.; Altman, R.B.; Eriksson, N.; Gage, B.F.; Kimmel, S.E.; Lee, M.T.M.; Limdi, N.A.; Page, D.; Roden, D.M.; Wagner, M.J.; et al. Estimation of the Warfarin Dose with Clinical and Pharmacogenetic Data. N. Engl. J. Med. 2009, 360, 753–764. [Google Scholar] [PubMed]
- Lenzini, P.; Wadelius, M.; Kimmel, S.; Anderson, J.L.; Jorgensen, A.L.; Pirmohamed, M.; Caldwell, M.D.; Limdi, N.; Burmester, J.K.; Dowd, M.B.; et al. Integration of genetic, clinical, and INR data to refine warfarin dosing. Clin. Pharmacol. Ther. 2010, 87, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Avery, P.J.; Jorgensen, A.; Hamberg, A.K.; Wadelius, M.; Pirmohamed, M.; Kamali, F. A proposal for an individualized pharmacogenetics-based warfarin initiation dose regimen for patients commencing anticoagulation therapy. Clin. Pharmacol. Ther. 2011, 90, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Sagreiya, H.; Berube, C.; Wen, A.; Ramakrishnan, R.; Mir, A.; Hamilton, A.; Altman, R.B. Extending and evaluating a warfarin dosing algorithm that includes CYP4F2 and pooled rare variants of CYP2C9. Pharmacogenet. Genom. 2010, 20, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Shahabi, P.; Scheinfeldt, L.B.; Lynch, D.E.; Schmidlen, T.J.; Perreault, S.; Keller, M.A.; Kasper, R.; Wawak, L.; Jarvis, J.P.; Gerry, N.P.; et al. An expanded pharmacogenomics warfarin dosing table with utility in generalised dosing guidance. Thromb. Haemost. 2016, 116, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Drozda, K.; Wong, S.; Patel, S.R.; Bress, A.P.; Nutescu, E.A.; Kittles, R.A.; Cavallari, L.H. Poor warfarin dose prediction with pharmacogenetic algorithms that exclude genotypes important for African Americans. Pharmacogenet. Genom. 2015, 25, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; On, Y.K.; Bang, O.Y.; Kim, J.W.; Huh, W.; Ko, J.W.; Kim, J.S.; Lee, S.Y. Development and comparison of a warfarin-dosing algorithm for Korean patients with atrial fibrillation. Clin. Ther. 2011, 33, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Ye, F.; Xie, D.; Zhu, Y.; Zhu, J.; Tao, Y.; Yu, F. A new algorithm to predict warfarin dose from polymorphisms of CYP4F2, CYP2C9 and VKORC1 and clinical variables: Derivation in Han Chinese patients with non valvular atrial fibrillation. Thromb. Haemost. 2012, 107, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Krishna Kumar, D.; Shewade, D.G.; Loriot, M.A.; Beaune, P.; Balachander, J.; Sai Chandran, B.V.; Adithan, C. Effect of CYP2C9, VKORC1, CYP4F2 and GGCX genetic variants on warfarin maintenance dose and explicating a new pharmacogenetic algorithm in South Indian population. Eur. J. Clin. Pharmacol. 2014, 70, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.C.; Marcatto, L.R.; Duarte, N.E.; Gadi Soares, R.A.; Cassaro Strunz, C.M.; Scanavacca, M.; Krieger, J.E.; Pereira, A.C. Development of a pharmacogenetic-based warfarin dosing algorithm and its performance in Brazilian patients: Highlighting the importance of population-specific calibration. Pharmacogenomics 2015, 16, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Biss, T.T.; Avery, P.J.; Brandao, L.R.; Chalmers, E.A.; Williams, M.D.; Grainger, J.D.; Leathart, J.B.; Hanley, J.P.; Daly, A.K.; Kamali, F. VKORC1 and CYP2C9 genotype and patient characteristics explain a large proportion of the variability in warfarin dose requirement among children. Blood 2012, 119, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.; Bajolle, F.; Siguret, V.; Lasne, D.; Golmard, J.L.; Elie, C.; Beaune, P.; Cheurfi, R.; Bonnet, D.; Loriot, M.A. Vitamin K antagonists in children with heart disease: Height and VKORC1 genotype are the main determinants of the warfarin dose requirement. Blood 2012, 119, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Caraco, Y.; Blotnick, S.; Muszkat, M. CYP2C9 genotype-guided warfarin prescribing enhances the efficacy and safety of anticoagulation: A prospective randomized controlled study. Clin. Pharmacol. Ther. 2008, 83, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Epstein, R.S.; Moyer, T.P.; Aubert, R.E.; DJ, O.K.; Xia, F.; Verbrugge, R.R.; Gage, B.F.; Teagarden, J.R. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J. Am. Coll. Cardiol. 2010, 55, 2804–2812. [Google Scholar] [CrossRef] [PubMed]
- McMillin, G.A.; Melis, R.; Wilson, A.; Strong, M.B.; Wanner, N.A.; Vinik, R.G.; Peters, C.L.; Pendleton, R.C. Gene-based warfarin dosing compared with standard of care practices in an orthopedic surgery population: A prospective, parallel cohort study. Ther. Drug Monit. 2010, 32, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Burmester, J.K.; Berg, R.L.; Yale, S.H.; Rottscheit, C.M.; Glurich, I.E.; Schmelzer, J.R.; Caldwell, M.D. A randomized controlled trial of genotype-based Coumadin initiation. Genet. Med. 2011, 13, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Gong, L.; Whirl-Carrillo, M.; Gage, B.F.; Scott, S.A.; Stein, C.M.; Anderson, J.L.; Kimmel, S.E.; Lee, M.T.; Pirmohamed, M.; et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin. Pharmacol. Ther. 2011, 90, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Horne, B.D.; Stevens, S.M.; Woller, S.C.; Samuelson, K.M.; Mansfield, J.W.; Robinson, M.; Barton, S.; Brunisholz, K.; Mower, C.P.; et al. A randomized and clinical effectiveness trial comparing two pharmacogenetic algorithms and standard care for individualizing warfarin dosing (CoumaGen-II). Circulation 2012, 125, 1997–2005. [Google Scholar] [CrossRef] [PubMed]
- Pirmohamed, M.; Burnside, G.; Eriksson, N.; Jorgensen, A.L.; Toh, C.H.; Nicholson, T.; Kesteven, P.; Christersson, C.; Wahlstrom, B.; Stafberg, C.; et al. A randomized trial of genotype-guided dosing of warfarin. N. Engl. J. Med. 2013, 369, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, S.E.; French, B.; Kasner, S.E.; Johnson, J.A.; Anderson, J.L.; Gage, B.F.; Rosenberg, Y.D.; Eby, C.S.; Madigan, R.A.; McBane, R.B.; et al. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N. Engl. J. Med. 2013, 369, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Pirmohamed, M.; Kamali, F.; Daly, A.K.; Wadelius, M. Oral anticoagulation: A critique of recent advances and controversies. Trends Pharmacol. Sci. 2015, 36, 153–163. [Google Scholar] [CrossRef] [PubMed]
- French, B.; Wang, L.; Gage, B.F.; Horenstein, R.B.; Limdi, N.A.; Kimmel, S.E. A systematic analysis and comparison of warfarin initiation strategies. Pharmacogenet. Genom. 2016, 26, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Gage, B.F.; Bass, A.R.; Lin, H.; Woller, S.C.; Stevens, S.M.; Al-Hammadi, N.; Li, J.; Rodriguez, T., Jr.; Miller, J.P.; McMillin, G.A.; et al. Effect of Genotype-Guided Warfarin Dosing on Clinical Events and Anticoagulation Control Among Patients Undergoing Hip or Knee Arthroplasty: The GIFT Randomized Clinical Trial. JAMA 2017, 318, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Saffian, S.M.; Duffull, S.B.; Wright, D. Warfarin Dosing Algorithms Underpredict Dose Requirements in Patients Requiring ≥7 mg Daily: A Systematic Review and Meta-analysis. Clin. Pharmacol. Ther. 2017, 102, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Hylek, E.M.; Evans-Molina, C.; Shea, C.; Henault, L.E.; Regan, S. Major hemorrhage and tolerability of warfarin in the first year of therapy among elderly patients with atrial fibrillation. Circulation 2007, 115, 2689–2696. [Google Scholar] [CrossRef] [PubMed]
- Pirmohamed, M.; James, S.; Meakin, S.; Green, C.; Scott, A.K.; Walley, T.J.; Farrar, K.; Park, B.K.; Breckenridge, A.M. Adverse drug reactions as cause of admission to hospital: Prospective analysis of 18 820 patients. BMJ 2004, 329, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Mega, J.L.; Walker, J.R.; Ruff, C.T.; Vandell, A.G.; Nordio, F.; Deenadayalu, N.; Murphy, S.A.; Lee, J.; Mercuri, M.F.; Giugliano, R.P.; et al. Genetics and the clinical response to warfarin and edoxaban: Findings from the randomised, double-blind ENGAGE AF-TIMI 48 trial. Lancet 2015, 385, 2280–2287. [Google Scholar] [CrossRef]
- Van Driest, S.L.; Shi, Y.; Bowton, E.A.; Schildcrout, J.S.; Peterson, J.F.; Pulley, J.; Denny, J.C.; Roden, D.M. Clinically actionable genotypes among 10,000 patients with preemptive pharmacogenomic testing. Clin. Pharmacol. Ther. 2014, 95, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Klein, T.E. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharmacol. Ther. 2011, 89, 464–467. [Google Scholar] [CrossRef] [PubMed]
- CPIC. Genes-Drug. Available online: https://cpicpgx.org/genes-drugs/ (accessed on 31 October 2017).
- Scott, S.A. Personalizing medicine with clinical pharmacogenetics. Genet. Med. 2011, 13, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Shirts, B.H.; Pritchard, C.C.; Walsh, T. Family-Specific Variants and the Limits of Human Genetics. Trends Mol. Med. 2016, 22, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.S.; Tabor, H.K.; Johnson, A.D.; Snively, B.M.; Assimes, T.L.; Auer, P.L.; Ioannidis, J.P.; Peters, U.; Robinson, J.G.; Sucheston, L.E.; et al. Quantifying rare, deleterious variation in 12 human cytochrome P450 drug-metabolism genes in a large-scale exome dataset. Hum. Mol. Genet. 2014, 23, 1957–1963. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M. Parlez-vous VUS? Genome Res. 2015, 25, 1423–1426. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Starita, L.M.; Ahituv, N.; Dunham, M.J.; Kitzman, J.O.; Roth, F.P.; Seelig, G.; Shendure, J.; Fowler, D.M. Variant Interpretation: Functional Assays to the Rescue. Am. J. Hum. Genet. 2017, 101, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Stephany, J.J.; Fields, S. Measuring the activity of protein variants on a large scale using deep mutational scanning. Nat. Protoc. 2014, 9, 2267–2284. [Google Scholar] [CrossRef] [PubMed]
- Gasperini, M.; Starita, L.; Shendure, J. The power of multiplexed functional analysis of genetic variants. Nat. Protoc. 2016, 11, 1782–1787. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Fowler, D.M.; Starita, L.M.; Haendel, M.A.; MacArthur, D.G.; Biesecker, L.G.; Worthey, E.; Chisholm, R.L.; Green, E.D.; Jacob, H.J.; et al. Bedside Back to Bench: Building Bridges between Basic and Clinical Genomic Research. Cell 2017, 169, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Krauss, R.M.; Roden, D.M.; Klein, T.E.; Fowler, D.M.; Terada, N.; Lin, L.; Riel-Mehan, M.; Do, T.P.; Kubo, M.; et al. New Pharmacogenomics Research Network: An Open Community Catalyzing Research and Translation in Precision Medicine. Clin. Pharmacol. Ther. 2017, 102, 897–902. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug Class | Drugs |
|---|---|
| Anticoagulants | Acenocoumarol, phenprocoumon, S-warfarin |
| Antihypertensives | Irbesartan, losartan |
| NSAIDs | Celecoxib, diclofenac, etodolac, ibuprofen, indomethacin, lornoxicam, mefenamic acid, suprofen, tenoxicam |
| Oral hypoglycemic agents | Chlorpropamide, glibenclamide, gliclazide, glimepiride, nateglinide, tolbutamide |
| Miscellaneous | Bosentan, fluvastatin, mestranol, phenytoin, torsemide |
| SNP | Effect | *Allele | Sequence Change Europeans | Overall Frequency Worldwide | European Frequency | African Frequency | East Asian Frequency | South Asian Frequency | Effect |
|---|---|---|---|---|---|---|---|---|---|
| rs1799853 | p.Arg144Cys | *2 | c.430C>T | 0.0914 | 0.1268 | 0.0235 | 0.0003 | 0.046 | PolyPhen: probably damaging; SIFT:tolerated; other:impaired S-warfarin metabolism in vitro [77] and decreased dose in vivo [82] |
| rs1057910 | p.Ile359Leu | *3 | c.1075A>C | 0.0637 | 0.0688 | 0.0126 | 0.0338 | 0.1131 | PolyPhen: possibly damaging; SIFT: deleterious; other: impaired S-warfarin and tolbutamide metabolism in vitro [78,79] and decreased warfarin dose in vivo [82] |
| rs2256871 | p.His251Arg | *9 | c.752A>G | 0.0067 | 0.0002 | 0.0754 | 0.0001 | 0.0001 | PolyPhen: probably damaging; SIFT: deleterious; other: no effect on phenytoin clearance in vivo [90] |
| rs7900194 | p.Arg150His | *8 | c.449G>A | 0.0052 | 0.0003 | 0.056 | 0.0001 | 0.0006 | PolyPhen:benign; SIFT:tolerated; other: increased activity towards tolbutamide in vitro [91], decreased activity towards phenytoin in vivo [90], decreased warfarin activity in vitro and in vivo [92] |
| rs28371685 | p.Arg335Trp | *11 | c.1003C>T | 0.0038 | 0.0021 | 0.0214 | 0.0001 | 0.0019 | PolyPhen: probably damaging; SIFT: deleterious; other: decreased activity towards warfarin in vivo and in vitro [87,91] |
| rs72558189 | p.Arg125His | *14 | c.374G>A | 0.003 | 0.0001 | >0.0001 | 0.0001 | 0.0204 | PolyPhen:benign; SIFT:deleterious; other: very low activity in vitro towards tolbutamide and warfarin [93] |
| rs9332239 | p.Pro489Ser | *12 | c.1465C>T | 0.0019 | 0.003 | 0.0006 | 0 | 0.0002 | PolyPhen: possibly damaging; SIFT: deleterious; other: decreased warfarin dose requirement [88] |
| rs2837168 | p.Asp360Glu | *5 | c.1080C>G | 0.0012 | <0.0001 | 0.0127 | 0 | 0 | PolyPhen: probably damaging; SIFT: deleterious; other: decreased activity towards warfarin and diclofenac in vitro [94]; decreased phenytoin clearance in vivo [90] |
| rs9332131 | p.Lys273Arg (fsTer34) | *6 | c.818delA | 0.0009 | <0.0001 | 0.0105 | 0 | 0 | Frameshift so inactivating; other: impaired phenytoin clearance in vivo [95] |
| rs182132442 | p.Pro279Thr | *29 | c.835C>A | 0.0004 | 0.0005 * | 0 | 0.0016 | 0.0001 | PolyPhen:benign; SIFT:tolerated; other: decreased activity in vitro with tolbutamide [89]; decreased warfarin clearance in vitro [93] |
| rs72558192 | p.Thr299Ala | *52 | c.895A>G | 0.0002 | 0 | 0 | 0.0035 | 0 | PolyPhen: probably damaging; SIFT: deleterious; other: mutation of known active residue based on crystal structures of flurbiprofen- and warfarin-bound CYP2C9 [96], decreased activity in vitro with tolbutamide [97] |
| rs72558187 | p.Leu90Pro | *13 | c.269T>C | 0.0001 | 0 | 0 | 0.002 | 0 | PolyPhen:benign; SIFT:tolerated; other: decreased activity in vitro with tolbutamide [97] |
| rs7900194 | p.Arg150Leu | *27 | c.449G>T | 0.0001 | 0 | 0 | 0.0017 | >0.0001 | PolyPhen:benign; SIFT:tolerated; other: decreased activity in vitro with tolbutamide [97]; decreased warfarin clearance in vitro [93] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daly, A.K.; Rettie, A.E.; Fowler, D.M.; Miners, J.O. Pharmacogenomics of CYP2C9: Functional and Clinical Considerations. J. Pers. Med. 2018, 8, 1. https://doi.org/10.3390/jpm8010001
Daly AK, Rettie AE, Fowler DM, Miners JO. Pharmacogenomics of CYP2C9: Functional and Clinical Considerations. Journal of Personalized Medicine. 2018; 8(1):1. https://doi.org/10.3390/jpm8010001
Chicago/Turabian StyleDaly, Ann K., Allan E. Rettie, Douglas M. Fowler, and John O. Miners. 2018. "Pharmacogenomics of CYP2C9: Functional and Clinical Considerations" Journal of Personalized Medicine 8, no. 1: 1. https://doi.org/10.3390/jpm8010001