
Identifying Proteins and Amino Acids Associated with Liver Cancer Risk: A Study Utilizing Mendelian Randomization and Bulk RNA Sequencing Analysis
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design of MR Analysis
2.2. PLC, Proteome, and Metabolome Sample
2.3. IV Selection
2.4. MR Analysis
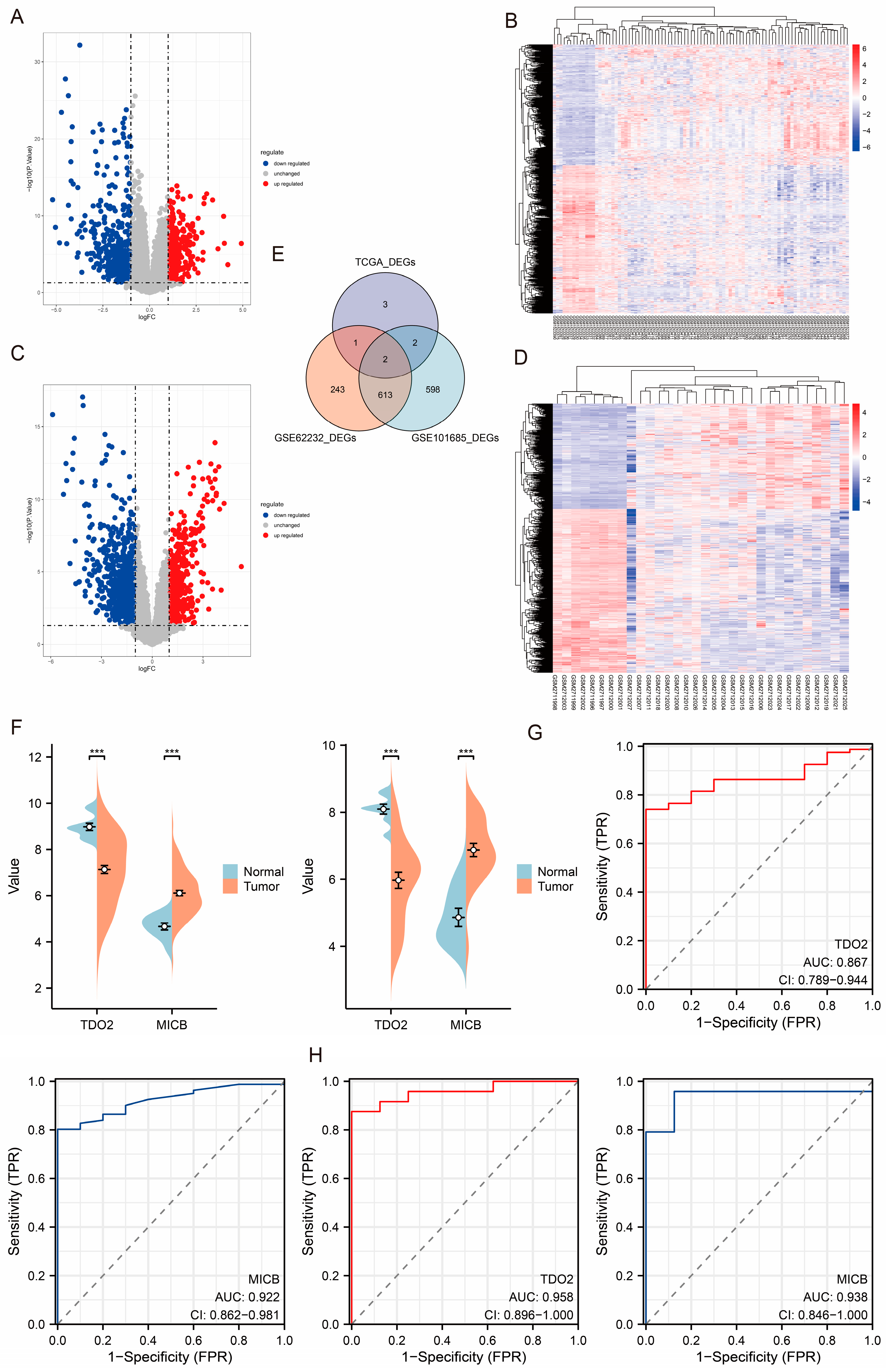
2.5. Identification of Differentially Expressed Genes (DEGs)
2.6. Enrichment Analysis
2.7. Analysis of Immune Infiltration
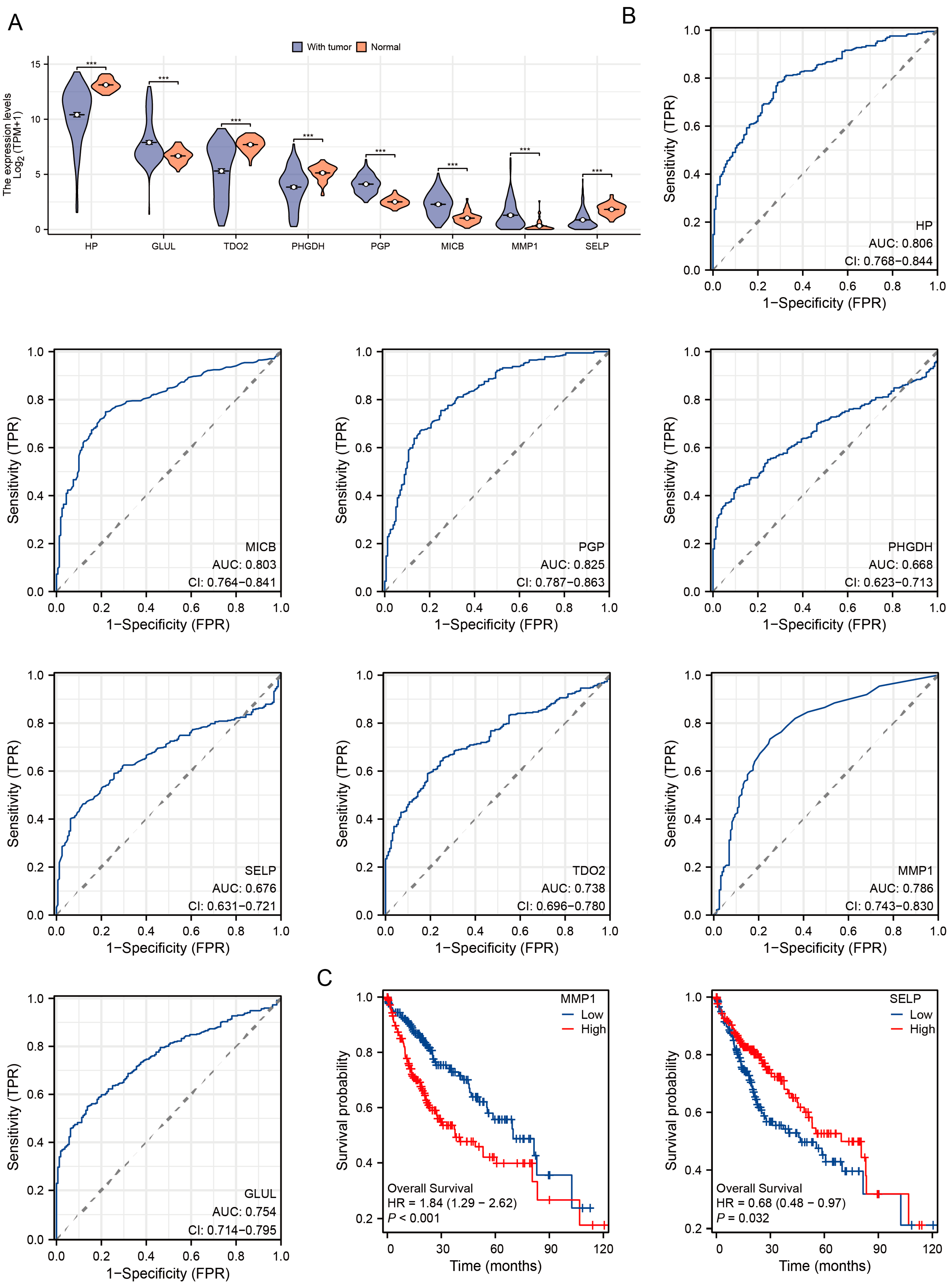
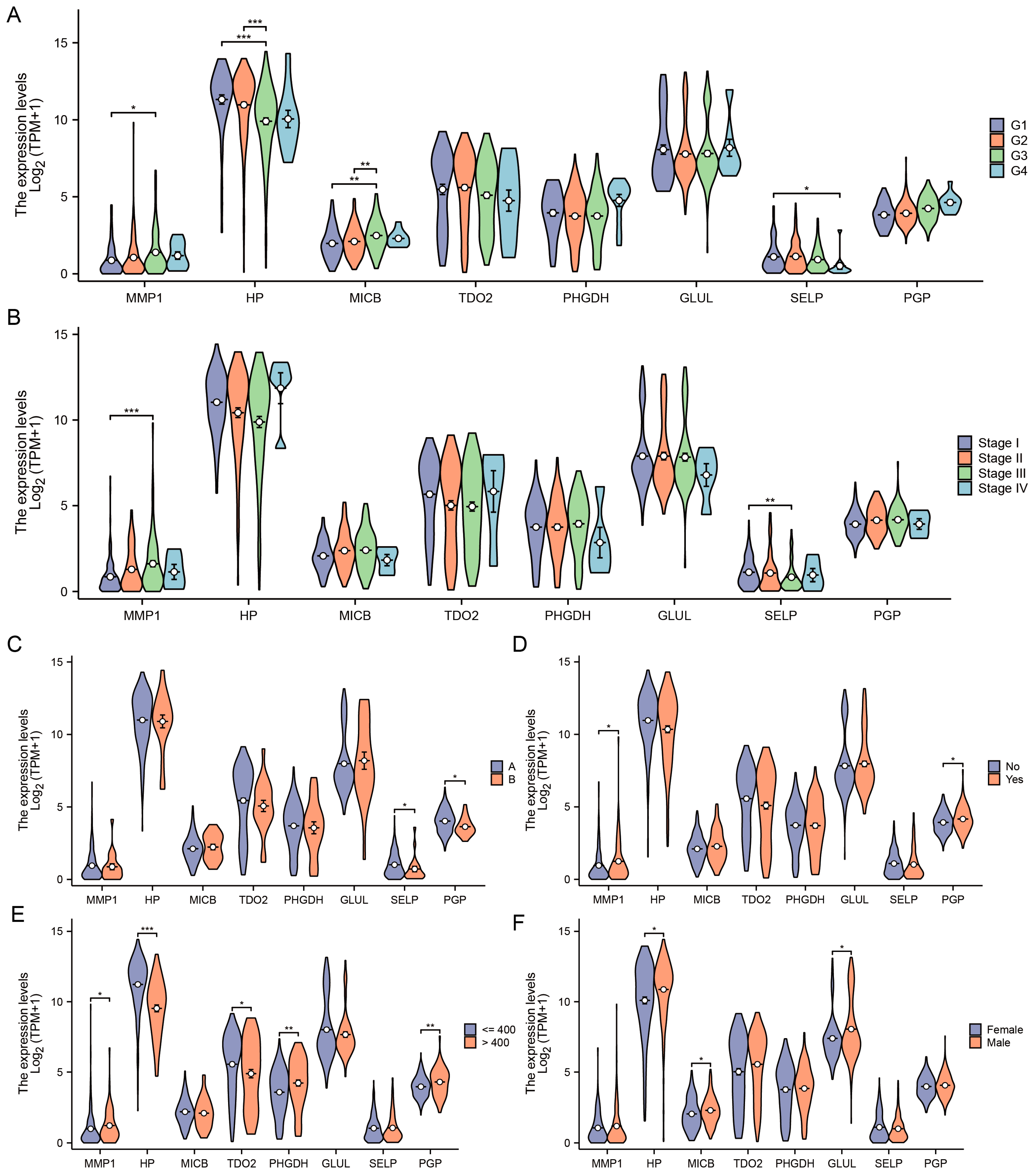
2.8. Diagnosis and Prognosis Analysis
3. Results
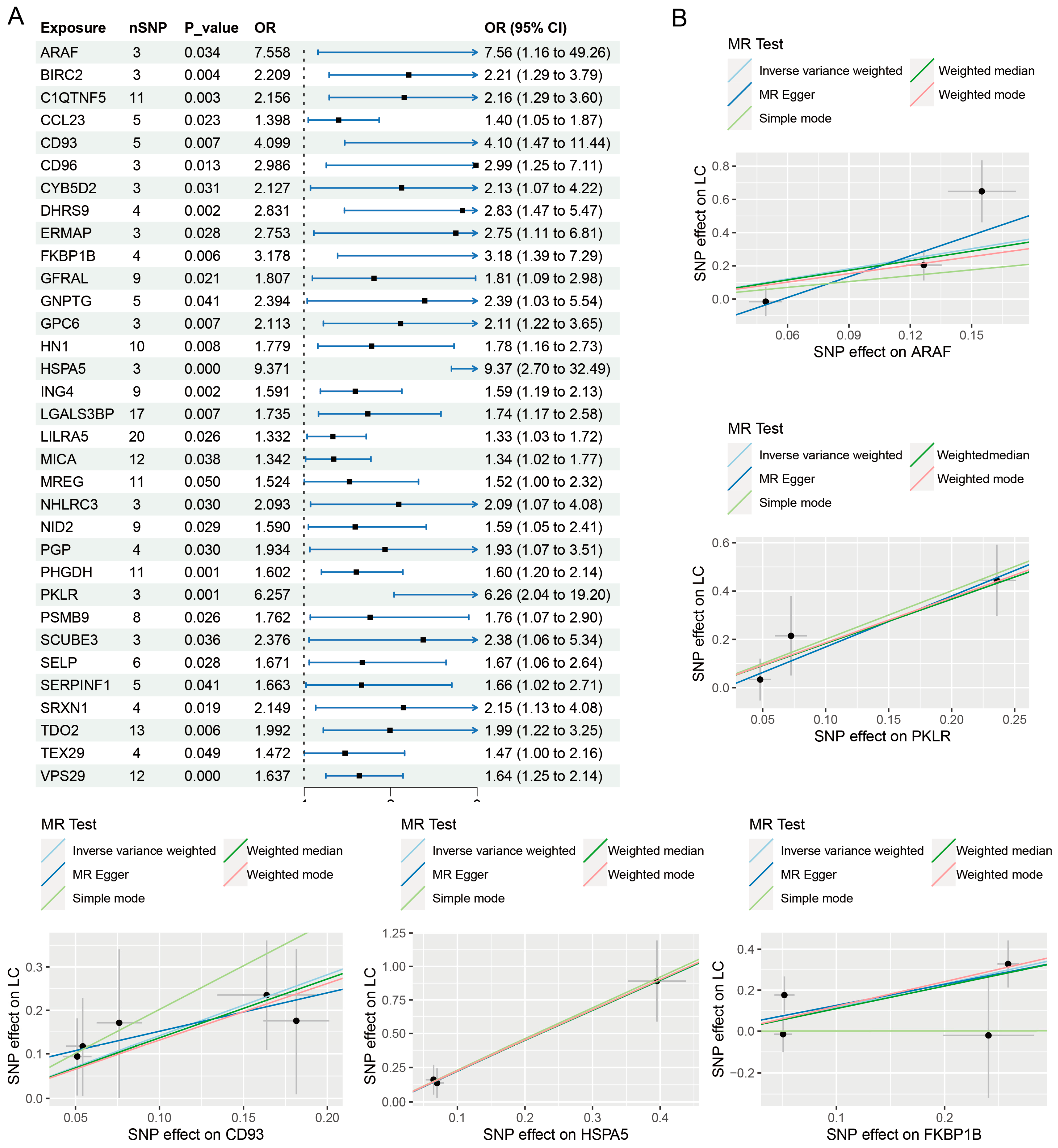
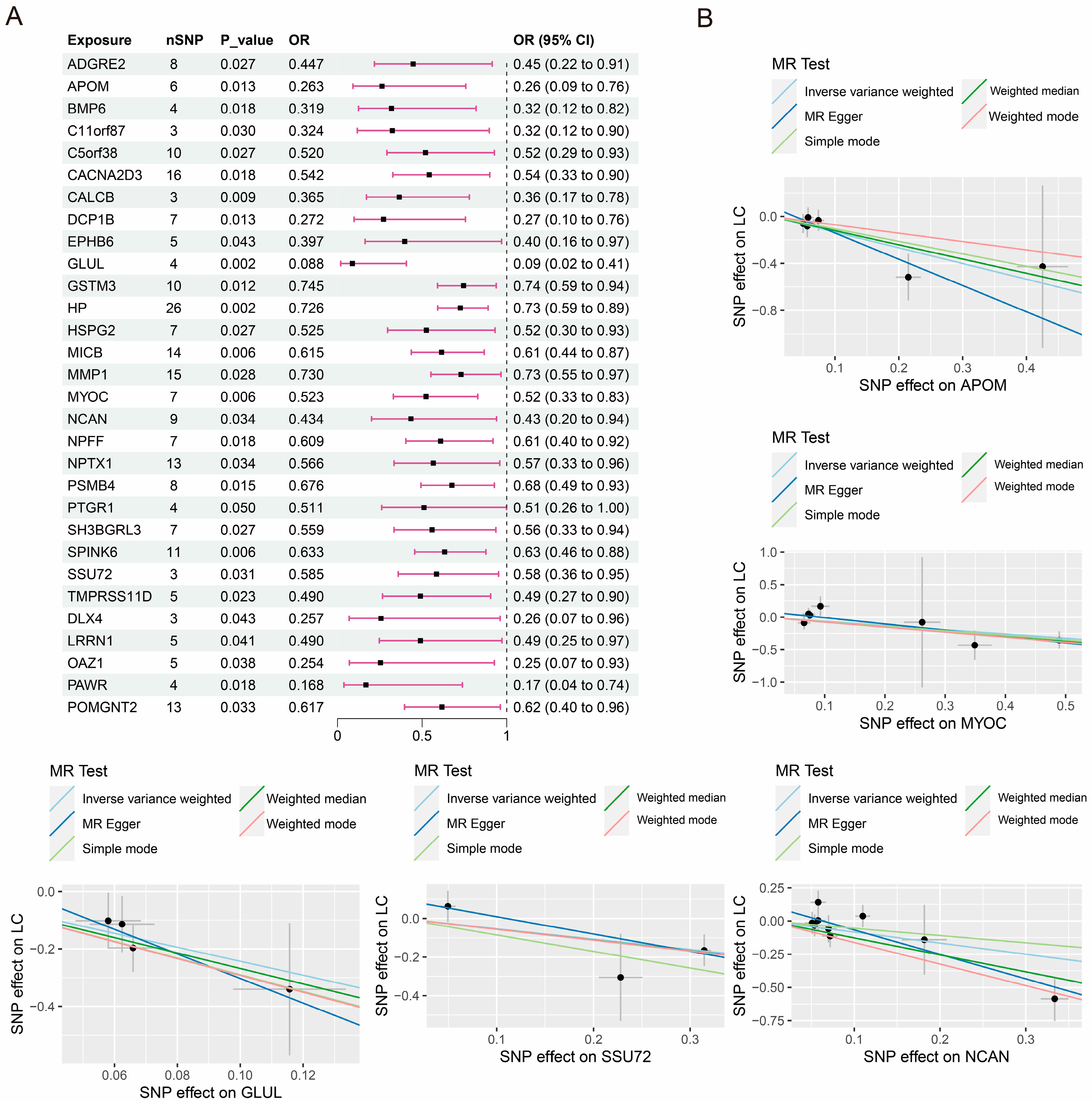
3.1. Causal Effects of Proteome on PLC
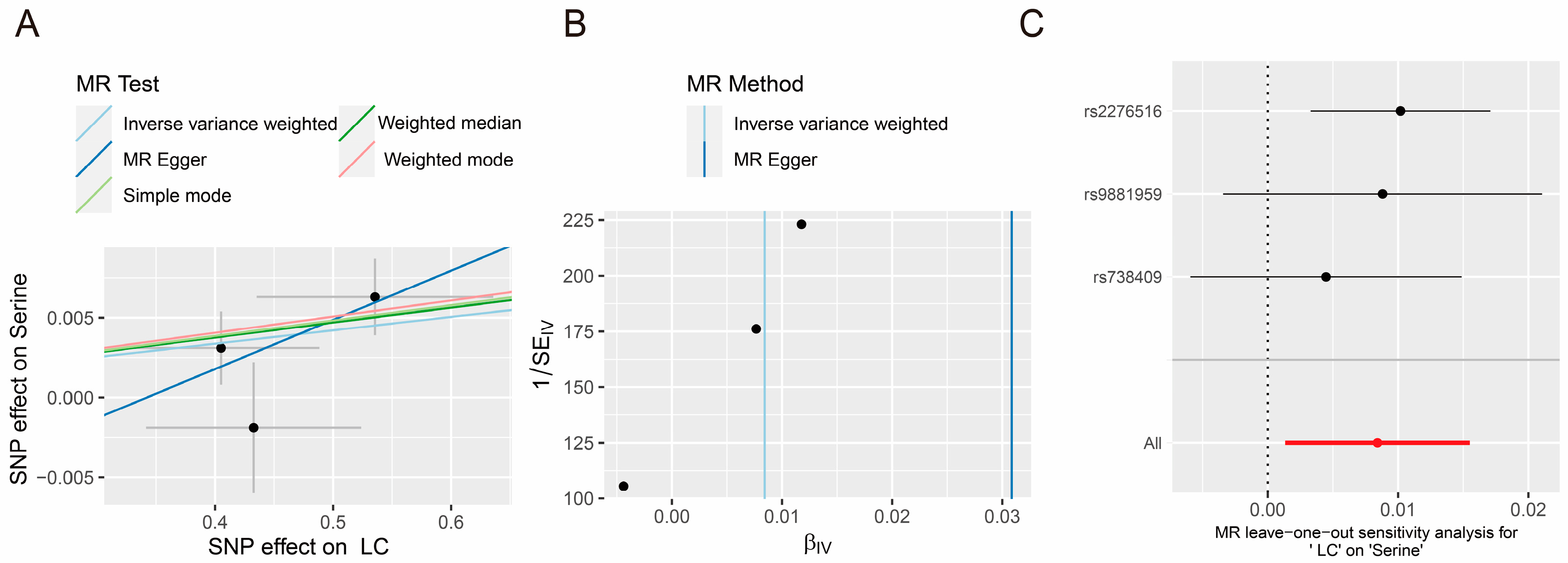
3.2. Causal Effects of Amino Acid and PLC
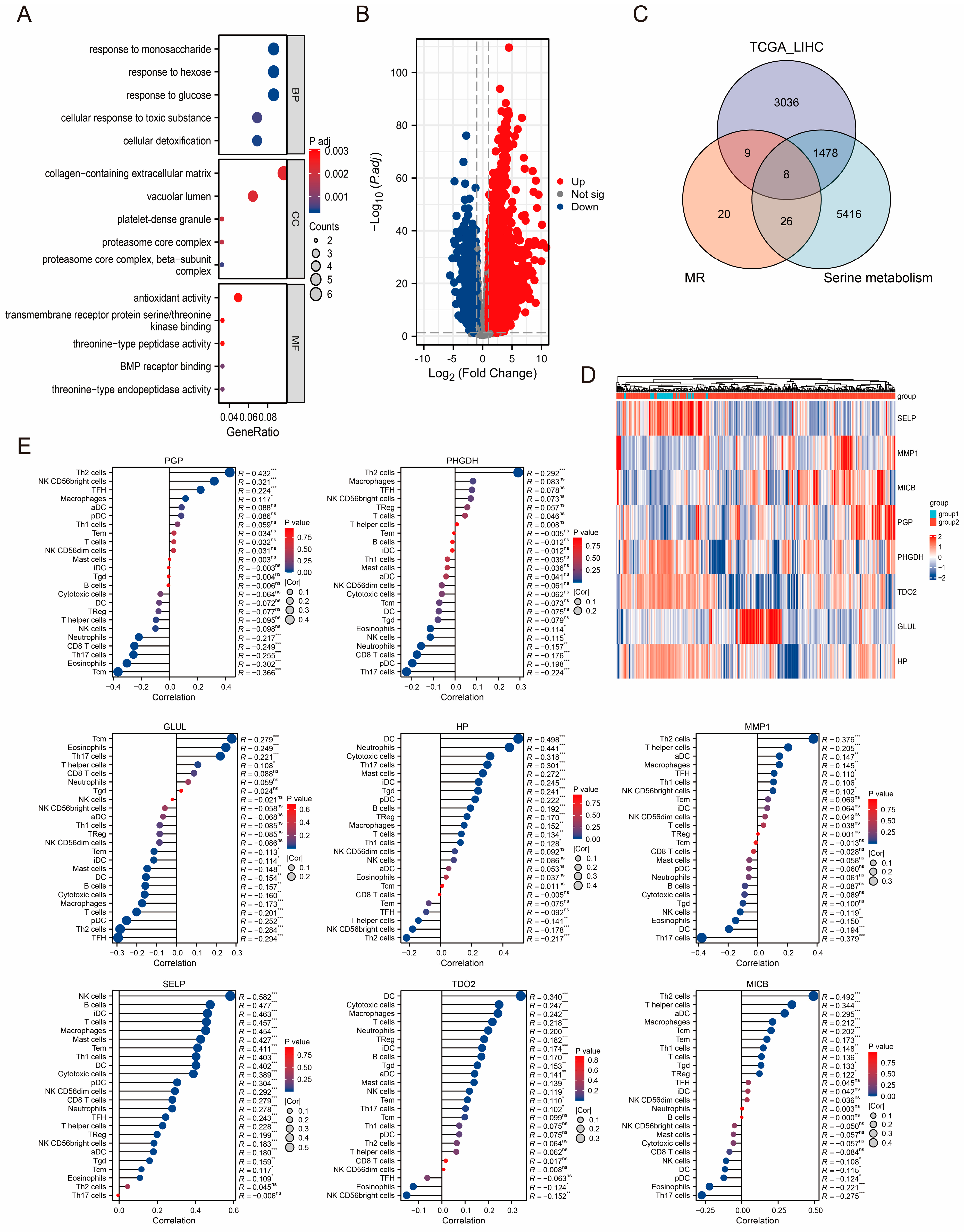
3.3. Identification of TCGA-LIHC Cohort-Related DEGs and Enrichment Analysis
3.4. Diagnostic and Prognostic Value of DEGs in TCGA-LIHC Cohort
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Wong, G.; Anstee, Q.M.; Henry, L. The Global Burden of Liver Disease. Clin. Gastroenterol. Hepatol. 2023, 21, 1978–1991. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Wang, J.J.; Luu, M.; Noureddin, M.; Kosari, K.; Agopian, V.G.; Rich, N.E.; Lu, S.C.; Tseng, H.R.; Nissen, N.N.; et al. The Mortality and Overall Survival Trends of Primary Liver Cancer in the United States. J. Natl. Cancer Inst. 2021, 113, 1531–1541. [Google Scholar] [CrossRef]
- Xie, D.Y.; Zhu, K.; Ren, Z.G.; Zhou, J.; Fan, J.; Gao, Q. A review of 2022 Chinese clinical guidelines on the management of hepatocellular carcinoma: Updates and insights. Hepatobiliary Surg. Nutr. 2023, 12, 216–228. [Google Scholar] [CrossRef]
- Zelber-Sagi, S.; Noureddin, M.; Shibolet, O. Lifestyle and Hepatocellular Carcinoma What Is the Evidence and Prevention Recommendations. Cancers 2021, 14, 103. [Google Scholar] [CrossRef]
- Stella, L.; Santopaolo, F.; Gasbarrini, A.; Pompili, M.; Ponziani, F.R. Viral hepatitis and hepatocellular carcinoma: From molecular pathways to the role of clinical surveillance and antiviral treatment. World J. Gastroenterol. 2022, 28, 2251–2281. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Wong, N.; Lam, W.; Kuang, M. Personalized treatment for hepatocellular carcinoma: Current status and future perspectives. J. Gastroenterol. Hepatol. 2022, 37, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Mato, J.M.; Elortza, F.; Lu, S.C.; Brun, V.; Paradela, A.; Corrales, F.J. Liver cancer-associated changes to the proteome: What deserves clinical focus. Expert. Rev. Proteom. 2018, 15, 749–756. [Google Scholar] [CrossRef]
- Berkemeyer, S. Primary Liver Cancers: Connecting the Dots of Cellular Studies and Epidemiology with Metabolomics. Int. J. Mol. Sci. 2023, 24, 2409. [Google Scholar] [CrossRef]
- Shang, S.; Plymoth, A.; Ge, S.; Feng, Z.; Rosen, H.R.; Sangrajrang, S.; Hainaut, P.; Marrero, J.A.; Beretta, L. Identification of osteopontin as a novel marker for early hepatocellular carcinoma. Hepatology 2012, 55, 483–490. [Google Scholar] [CrossRef]
- Du, Z.; Liu, X.; Wei, X.; Luo, H.; Li, P.; Shi, M.; Guo, B.; Cui, Y.; Su, Z.; Zeng, J.; et al. Quantitative proteomics identifies a plasma multi-protein model for detection of hepatocellular carcinoma. Sci. Rep. 2020, 10, 15552. [Google Scholar] [CrossRef]
- Wei, T.; Liu, J.; Ma, S.; Wang, M.; Yuan, Q.; Huang, A.; Wu, Z.; Shang, D.; Yin, P. A Nucleotide Metabolism-Related Gene Signature for Risk Stratification and Prognosis Prediction in Hepatocellular Carcinoma Based on an Integrated Transcriptomics and Metabolomics Approach. Metabolites 2023, 13, 1116. [Google Scholar] [CrossRef]
- Katan, M.B. Apolipoprotein E isoforms, serum cholesterol, and cancer. Lancet 1986, 1, 507–508. [Google Scholar] [CrossRef] [PubMed]
- Ren, N.; Dai, S.; Ma, S.; Yang, F. Strategies for activity analysis of single nucleotide polymorphisms associated with human diseases. Clin. Genet. 2023, 103, 392–400. [Google Scholar] [CrossRef]
- Murphy, N.; Jenab, M.; Gunter, M.J. Adiposity and gastrointestinal cancers: Epidemiology, mechanisms and future directions. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 659–670. [Google Scholar] [CrossRef]
- Lawler, T.; Warren Andersen, S. Serum 25-Hydroxyvitamin D and Cancer Risk: A Systematic Review of Mendelian Randomization Studies. Nutrients 2023, 15, 422. [Google Scholar] [CrossRef]
- Li, M.; Wang, H.; Zhang, X.J.; Cai, J.; Li, H. NAFLD: An Emerging Causal Factor for Cardiovascular Disease. Physiology 2023, 38, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Z.Q.; Chen, Y.Z.; Huang, Z.M.; Luo, Y.H.; Zeng, J.J.; Wang, Y.; Tan, J.; Chen, Y.X.; Fang, J.Y. Metabolic syndrome, its components, and gastrointestinal cancer risk: A meta-analysis of 31 prospective cohorts and Mendelian randomization study. J. Gastroenterol. Hepatol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Shi, W.; Chen, K.; Lu, C.; Li, X.; Li, Q. Elucidating the causal association between gut microbiota and intrahepatic cholangiocarcinoma through Mendelian randomization analysis. Front. Microbiol. 2023, 14, 1288525. [Google Scholar] [CrossRef]
- Zhao, J.H.; Stacey, D.; Eriksson, N.; Macdonald-Dunlop, E.; Hedman, Å.K.; Kalnapenkis, A.; Enroth, S.; Cozzetto, D.; Digby-Bell, J.; Marten, J.; et al. Genetics of circulating inflammatory proteins identifies drivers of immune-mediated disease risk and therapeutic targets. Nat. Immunol. 2023, 24, 1540–1551. [Google Scholar] [CrossRef]
- Ning, Z.; Huang, Y.; Lu, H.; Zhou, Y.; Tu, T.; Ouyang, F.; Liu, Y.; Liu, Q. Novel Drug Targets for Atrial Fibrillation Identified Through Mendelian Randomization Analysis of the Blood Proteome. Cardiovasc. Drugs Ther. 2023. [Google Scholar] [CrossRef]
- Kimhofer, T.; Fye, H.; Taylor-Robinson, S.; Thursz, M.; Holmes, E. Proteomic and metabonomic biomarkers for hepatocellular carcinoma: A comprehensive review. Br. J. Cancer 2015, 112, 1141–1156. [Google Scholar] [CrossRef] [PubMed]
- Skrivankova, V.W.; Richmond, R.C.; Woolf, B.; Yarmolinsky, J.; Davies, N.M.; Swanson, S.A.; VanderWeele, T.J.; Higgins, J.; Timpson, N.J.; Dimou, N.; et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE-MR Statement. JAMA 2021, 326, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Bourgault, J.; Abner, E.; Manikpurage, H.D.; Pujol-Gualdo, N.; Laisk, T.; Gobeil, É.; Gagnon, E.; Girard, A.; Mitchell, P.L.; Thériault, S.; et al. Proteome-Wide Mendelian Randomization Identifies Causal Links Between Blood Proteins and Acute Pancreatitis. Gastroenterology 2023, 164, 953–965.e3. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.L.; Burgess, S. Efficient design for Mendelian randomization studies: Subsample and 2-sample instrumental variable estimators. Am. J. Epidemiol. 2013, 178, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Greco M, F.D.; Minelli, C.; Sheehan, N.A.; Thompson, J.R. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat. Med. 2015, 34, 2926–2940. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef]
- Lu, Y.; Huang, C.; Gao, L.; Xu, Y.J.; Chia, S.E.; Chen, S.; Li, N.; Yu, K.; Ling, Q.; Cheng, Q.; et al. Identification of serum biomarkers associated with hepatitis B virus-related hepatocellular carcinoma and liver cirrhosis using mass-spectrometry-based metabolomics. Metabolomics 2015, 11, 1526–1538. [Google Scholar] [CrossRef]
- Di Poto, C.; He, S.; Varghese, R.S.; Zhao, Y.; Ferrarini, A.; Su, S.; Karabala, A.; Redi, M.; Mamo, H.; Rangnekar, A.S.; et al. Identification of race-associated metabolite biomarkers for hepatocellular carcinoma in patients with liver cirrhosis and hepatitis C virus infection. PLoS ONE 2018, 13, e0192748. [Google Scholar] [CrossRef]
- Zou, R.C.; Xiao, S.F.; Shi, Z.T.; Ke, Y.; Tang, H.R.; Wu, T.G.; Guo, Z.T.; Ni, F.; Li, W.X.; Wang, L. Identification of metabolism-associated pathways and genes involved in male and female liver cancer patients. J. Theor. Biol. 2019, 480, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z. Identification of the Association between Hepatitis B Virus and Liver Cancer using Machine Learning Approaches based on Amino Acid. In Proceedings of the 2020 10th International Conference on Bioscience, Biochemistry and Bioinformatics, Kyoto Japan, 19–22 January 2020; pp. 56–63. [Google Scholar]
- Nwosu, Z.C.; Megger, D.A.; Hammad, S.; Sitek, B.; Roessler, S.; Ebert, M.P.; Meyer, C.; Dooley, S. Identification of the Consistently Altered Metabolic Targets in Human Hepatocellular Carcinoma. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 303–323.e1. [Google Scholar] [CrossRef] [PubMed]
- Chaerkady, R.; Harsha, H.C.; Nalli, A.; Gucek, M.; Vivekanandan, P.; Akhtar, J.; Cole, R.N.; Simmers, J.; Schulick, R.D.; Singh, S.; et al. A quantitative proteomic approach for identification of potential biomarkers in hepatocellular carcinoma. J. Proteome Res. 2008, 7, 4289–4298. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Mi, W.; Cai, J.; Ying, W.; Liu, F.; Lu, H.; Qiao, Y.; Jia, W.; Bi, X.; Lu, N.; et al. Quantitative proteomic signature of liver cancer cells: Tissue transglutaminase 2 could be a novel protein candidate of human hepatocellular carcinoma. J. Proteome Res. 2008, 7, 3847–3859. [Google Scholar] [CrossRef]
- Ozawa, H.; Hirayama, A.; Shoji, F.; Maruyama, M.; Suzuki, K.; Yamanaka-Okumura, H.; Tatano, H.; Morine, Y.; Soga, T.; Shimada, M.; et al. Comprehensive Dipeptide Analysis Revealed Cancer-Specific Profile in the Liver of Patients with Hepatocellular Carcinoma and Hepatitis. Metabolites 2020, 10, 442. [Google Scholar] [CrossRef] [PubMed]
- Uzzaman, A.; Zhang, X.; Qiao, Z.; Zhan, H.; Sohail, A.; Wahid, A.; Shang, Z.; Guan, X.; Cao, C.X.; Xiao, H. Discovery of small extracellular vesicle proteins from human serum for liver cirrhosis and liver cancer. Biochimie 2020, 177, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Hilakivi-Clarke, L.; Shaha, A.; Wang, Y.; Wang, X.; Deng, Y.; Lai, J.; Kang, N. Metabolic reprogramming and its clinical implication for liver cancer. Hepatology 2023, 78, 1602–1624. [Google Scholar] [CrossRef]
- Fan, C.; Kam, S.; Ramadori, P. Metabolism-Associated Epigenetic and Immunoepigenetic Reprogramming in Liver Cancer. Cancers 2021, 13, 5250. [Google Scholar] [CrossRef]
- Guo, H.; Xu, J.; Zheng, Q.; He, J.; Zhou, W.; Wang, K.; Huang, X.; Fan, Q.; Ma, J.; Cheng, J.; et al. NRF2 SUMOylation promotes de novo serine synthesis and maintains HCC tumorigenesis. Cancer Lett. 2019, 466, 39–48. [Google Scholar] [CrossRef]
- Du, D.; Liu, C.; Qin, M.; Zhang, X.; Xi, T.; Yuan, S.; Hao, H.; Xiong, J. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm. Sin. B 2022, 12, 558–580. [Google Scholar] [CrossRef]
- Labuschagne, C.F.; van den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xu, L.; Wu, G.; Huang, Z.; Huang, L.; Zhang, F.; Wei, C.; Shen, Q.; Li, R.; Zhang, L.; et al. Remodeling Serine Synthesis and Metabolism via Nanoparticles (NPs)-Mediated CFL1 Silencing to Enhance the Sensitivity of Hepatocellular Carcinoma to Sorafenib. Adv. Sci. 2023, 10, e2207118. [Google Scholar] [CrossRef]
- Wang, H.; Lin, F.; Xu, Z.; Yu, S.; Li, G.; Liao, S.; Zhao, W.; Zhang, F.; Wang, J.; Wang, S.; et al. ZEB1 Transcriptionally Activates PHGDH to Facilitate Carcinogenesis and Progression of HCC. Cell Mol. Gastroenterol. Hepatol. 2023, 16, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Z.X.; Wang, J.G.; Li, L.H.; Shen, W.L.; Dang, X.W. Deubiquitinating enzyme Josephin-2 stabilizes PHGDH to promote a cancer stem cell phenotype in hepatocellular carcinoma. Genes. Genom. 2023, 45, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Hao, Y.; Feng, J.; Liu, H.; Li, S.T.; Feng, J.; Jiang, Z.; Ye, L.; Zhou, Y.; Sun, Y.; et al. Non-canonical phosphoglycerate dehydrogenase activity promotes liver cancer growth via mitochondrial translation and respiratory metabolism. EMBO J. 2022, 41, e111550. [Google Scholar] [CrossRef] [PubMed]
- Trézéguet, V.; Fatrouni, H.; Merched, A.J. Immuno-Metabolic Modulation of Liver Oncogenesis by the Tryptophan Metabolism. Cells 2021, 10, 3469. [Google Scholar] [CrossRef]
- Stone, T.W.; Williams, R.O. Modulation of T cells by tryptophan metabolites in the kynurenine pathway. Trends Pharmacol. Sci. 2023, 44, 442–456. [Google Scholar] [CrossRef]
- Liu, H.; Xiang, Y.; Zong, Q.B.; Dai, Z.T.; Wu, H.; Zhang, H.M.; Huang, Y.; Shen, C.; Wang, J.; Lu, Z.X.; et al. TDO2 modulates liver cancer cell migration and invasion via the Wnt5a pathway. Int. J. Oncol. 2022, 60, 72. [Google Scholar] [CrossRef]
- Wu, Z.; Yan, L.; Lin, J.; Ke, K.; Yang, W. Constitutive TDO2 expression promotes liver cancer progression by an autocrine IL-6 signaling pathway. Cancer Cell Int. 2021, 21, 538. [Google Scholar] [CrossRef]
- Ferrari de Andrade, L.; Kumar, S.; Luoma, A.M.; Ito, Y.; Alves da Silva, P.H.; Pan, D.; Pyrdol, J.W.; Yoon, C.H.; Wucherpfennig, K.W. Inhibition of MICA and MICB Shedding Elicits NK-Cell-Mediated Immunity against Tumors Resistant to Cytotoxic T Cells. Cancer Immunol. Res. 2020, 8, 769–780. [Google Scholar] [CrossRef]
- Fang, L.; Gong, J.; Wang, Y.; Liu, R.; Li, Z.; Wang, Z.; Zhang, Y.; Zhang, C.; Song, C.; Yang, A.; et al. MICA/B expression is inhibited by unfolded protein response and associated with poor prognosis in human hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2014, 33, 76. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Kim, D.; Jung, J.; Kang, H.; Cho, H. NLRP3 Deficiency in Hepatocellular Carcinoma Enhances Surveillance of NK-92 through a Modulation of MICA/B. Int. J. Mol. Sci. 2021, 22, 9285. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exposure | Cochran’s Q Test | Horizontal Pleiotropy | MR-PRESSO | ||
|---|---|---|---|---|---|
| I2 | p-Value | Egger Intercept | p-Value | p-Value | |
| ADGRE2 | 0 | 0.973 | −0.024 | 0.742 | 0.976 |
| APOM | 0 | 0.766 | 0.083 | 0.377 | 0.682 |
| ARAF | 62% | 0.072 | −0.240 | 0.489 | NA |
| BIRC2 | 4% | 0.353 | −0.135 | 0.413 | 0.785 |
| BMP6 | 0 | 0.954 | 0.053 | 0.759 | 0.601 |
| C11orf87 | 0 | 0.924 | 0.007 | 0.961 | NA |
| C1QTNF5 | 16% | 0.294 | −0.061 | 0.491 | 0.304 |
| C5orf38 | 27% | 0.199 | 0.049 | 0.383 | 0.202 |
| CACNA2D3 | 2% | 0.425 | 0.019 | 0.702 | 0.524 |
| CALCB | 0 | 0.733 | 0.053 | 0.640 | 0.904 |
| CCL23 | 0 | 0.957 | −0.148 | 0.794 | 0.963 |
| CD93 | 0 | 0.967 | 0.062 | 0.596 | 0.994 |
| CD96 | 0 | 0.993 | −0.263 | 0.925 | 0.810 |
| CYB5D2 | 0 | 0.775 | 0.057 | 0.606 | 0.315 |
| DCP1B | 0 | 0.467 | 0.252 | 0.572 | 0.413 |
| DHRS9 | 0 | 0.400 | −0.090 | 0.441 | 0.447 |
| EPHB6 | 34% | 0.195 | 0.047 | 0.834 | 0.388 |
| ERMAP | 0 | 0.786 | −0.077 | 0.615 | 0.697 |
| FKBP1B | 13% | 0.329 | 0.023 | 0.851 | 0.539 |
| GFRAL | 0 | 0.495 | −0.069 | 0.418 | 0.606 |
| GLUL | 0 | 0.904 | 0.125 | 0.704 | 0.891 |
| GNPTG | 0 | 0.616 | −0.074 | 0.492 | 0.599 |
| GPC6 | 0 | 0.572 | −0.099 | 0.501 | 0.056 |
| GSTM3 | 0 | 0.568 | −0.109 | 0.168 | 0.588 |
| HN1 | 0 | 0.919 | 0.041 | 0.472 | 0.856 |
| HP | 0 | 0.830 | −0.013 | 0.771 | 0.817 |
| HSPA5 | 0 | 0.973 | −0.003 | 0.983 | NA |
| HSPG2 | 0 | 0.527 | −0.030 | 0.664 | 0.59 |
| ING4 | 0 | 0.740 | −0.022 | 0.754 | 0.688 |
| LGALS3BP | 21% | 0.213 | −0.034 | 0.467 | 0.247 |
| LILRA5 | 0 | 0.644 | 0.010 | 0.766 | 0.762 |
| MICA | 0 | 0.795 | 0.105 | 0.332 | 0.828 |
| MICB | 0 | 0.770 | 0.031 | 0.592 | 0.827 |
| MMP1 | 0 | 0.612 | −0.010 | 0.817 | 0.393 |
| MREG | 8% | 0.370 | −0.043 | 0.621 | 0.573 |
| MYOC | 0 | 0.489 | 0.088 | 0.218 | 0.439 |
| NCAN | 29% | 0.184 | 0.117 | 0.073 | 0.159 |
| NHLRC3 | 0 | 0.749 | −0.087 | 0.594 | 0.683 |
| NID2 | 0 | 0.497 | 0.022 | 0.769 | 0.542 |
| NPFF | 0 | 0.877 | −0.017 | 0.797 | 0.885 |
| NPTX1 | 0 | 0.983 | −0.005 | 0.918 | 0.985 |
| PGP | 0 | 0.886 | 0.048 | 0.893 | 0.764 |
| PHGDH | 0 | 0.718 | −0.047 | 0.591 | 0.712 |
| PKLR | 0 | 0.723 | −0.043 | 0.760 | NA |
| PSMB4 | 0 | 0.874 | −0.061 | 0.450 | 0.926 |
| PSMB9 | 0 | 0.492 | −0.027 | 0.686 | 0.633 |
| PTGR1 | 0 | 0.942 | −0.035 | 0.904 | 0.953 |
| SCUBE3 | 0 | 0.999 | 0.005 | 0.971 | NA |
| SELP | 0 | 0.919 | 0.071 | 0.469 | 0.467 |
| SERPINF1 | 0 | 0.450 | 0.014 | 0.922 | 0.515 |
| SH3BGRL3 | 3% | 0.400 | −0.033 | 0.657 | 0.615 |
| SPINK6 | 5% | 0.395 | −0.205 | 0.203 | 0.382 |
| SRXN1 | 0 | 0.997 | 0.019 | 0.934 | 0.925 |
| SSU72 | 0 | 0.396 | 0.098 | 0.497 | 0.824 |
| TDO2 | 0 | 0.809 | −0.058 | 0.422 | 0.721 |
| TEX29 | 0 | 0.687 | −0.443 | 0.447 | 0.534 |
| TMPRSS11D | 1% | 0.400 | 0.094 | 0.385 | 0.551 |
| VPS29 | 0 | 0.548 | −0.025 | 0.639 | 0.483 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, C.; Tang, L.; Yao, J.; Tan, G. Identifying Proteins and Amino Acids Associated with Liver Cancer Risk: A Study Utilizing Mendelian Randomization and Bulk RNA Sequencing Analysis. J. Pers. Med. 2024, 14, 262. https://doi.org/10.3390/jpm14030262
Ma C, Tang L, Yao J, Tan G. Identifying Proteins and Amino Acids Associated with Liver Cancer Risk: A Study Utilizing Mendelian Randomization and Bulk RNA Sequencing Analysis. Journal of Personalized Medicine. 2024; 14(3):262. https://doi.org/10.3390/jpm14030262
Chicago/Turabian StyleMa, Chi, Ling Tang, Jiaqi Yao, and Guang Tan. 2024. "Identifying Proteins and Amino Acids Associated with Liver Cancer Risk: A Study Utilizing Mendelian Randomization and Bulk RNA Sequencing Analysis" Journal of Personalized Medicine 14, no. 3: 262. https://doi.org/10.3390/jpm14030262