
The Role of Matrix Metalloproteinases in Hemorrhagic Transformation in the Treatment of Stroke with Tissue Plasminogen Activator
 ,
, 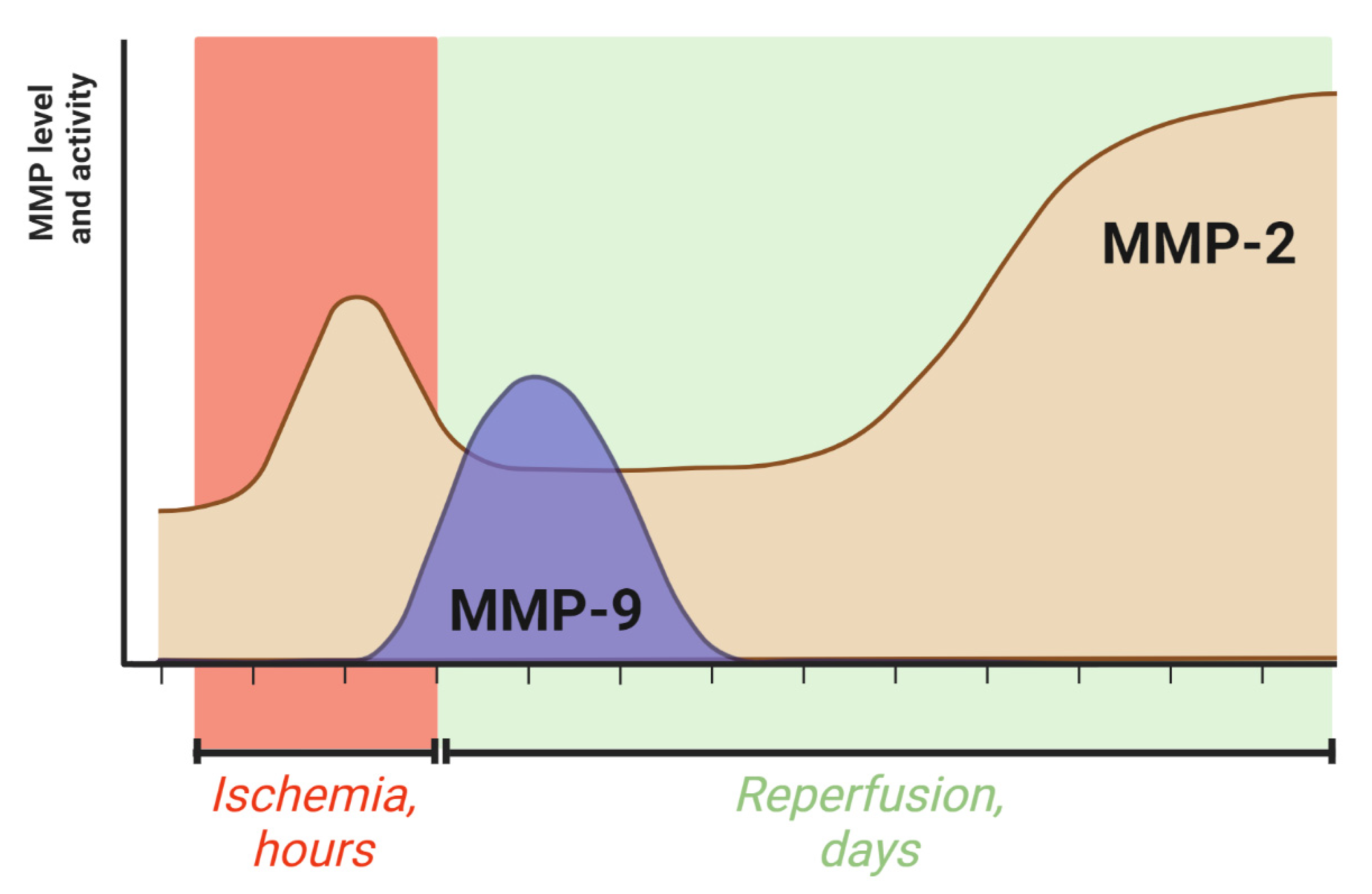{kind=link}
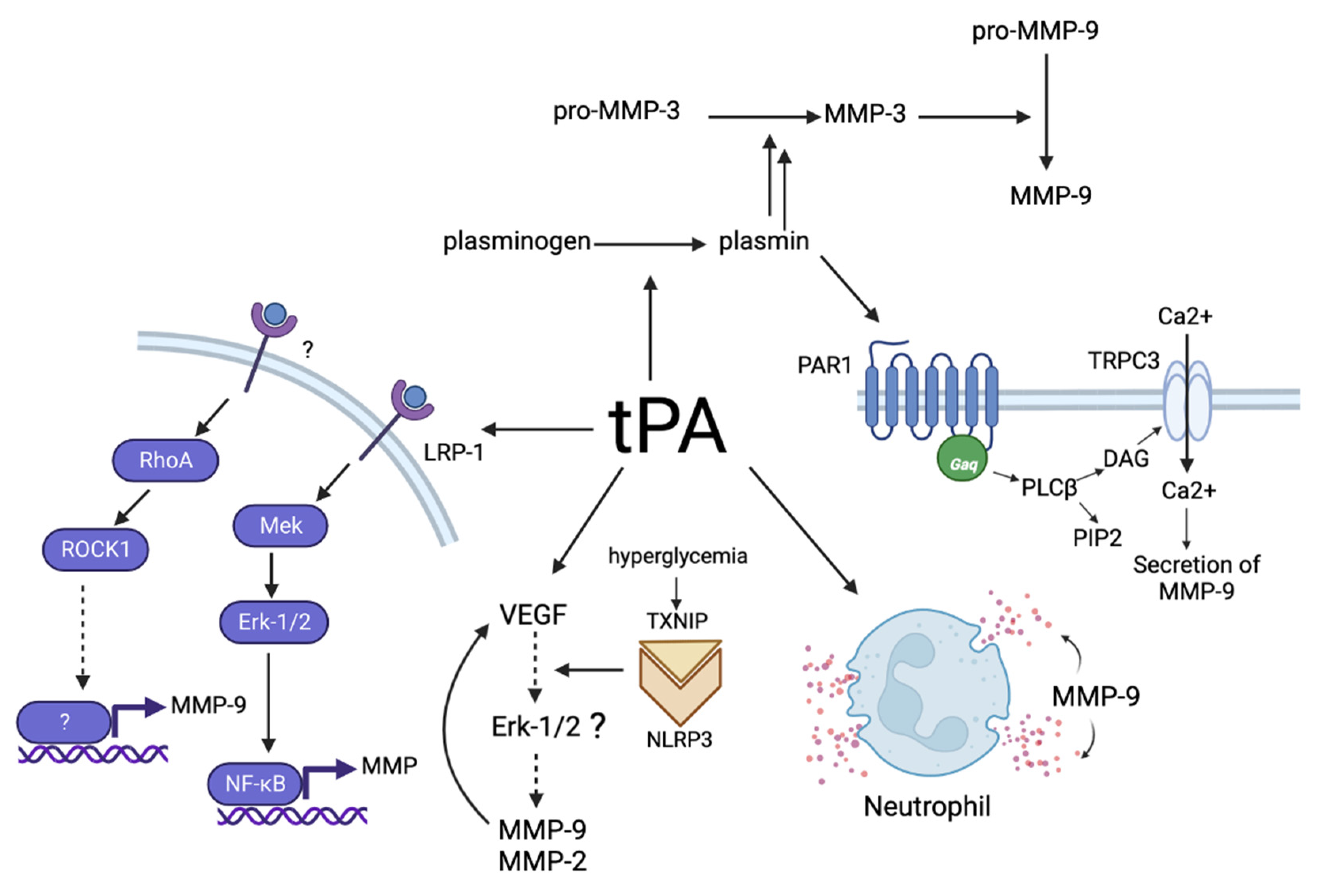{kind=link}
Abstract
:1. Introduction
2. Thrombolytic Therapy with Tissue Plasminogen Activator
3. Hemorrhagic Transformation and Activation of MMP in Ischemic Stroke
4. The Dynamics of MMP-2 and MMP-9 Activity during Ischemic Stroke
5. Mechanisms of the Influence of tPA on MMPs
6. Ways to Reduce MMP Activation in Ischemic Stroke
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.; Culebras, A.; Elkind, M.S.V.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An Updated Definition of Stroke for the 21st Century: A Statement for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2013, 396, 129–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, B.C.V.; Khatri, P. Stroke. Lancet 2020, 396, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Dewar, B.; Shamy, M. TPA for Acute Ischemic Stroke and Its Controversies: A Review. Neurohospitalist 2020, 10, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. 2018 Guidelines for the Early Management of Patients with Acute Ischemic Stroke: A Guideline for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2018, 49, e46–e110. [Google Scholar] [CrossRef]
- Honig, A.; Percy, J.; Sepehry, A.A.; Gomez, A.G.; Field, T.S.; Benavente, O.R. Hemorrhagic Transformation in Acute Ischemic Stroke: A Quantitative Systematic Review. J. Clin. Med. 2022, 11, 1162. [Google Scholar] [CrossRef]
- NINDS rt-PA Stroke Study Group. NINDS RTPA: Tissue Plasminogen Activator for Acute Ischemic Stroke. N. Engl. J. Med. 1995, 333, 1581–1587. [Google Scholar] [CrossRef]
- Chapman, S.N.; Mehndiratta, P.; Johansen, M.C.; McMurry, T.L.; Johnston, K.C.; Southerland, A.M. Current Perspectives on the Use of Intravenous Recombinant Tissue Plasminogen Activator (TPA) for Treatment of Acute Ischemic Stroke. Vasc. Health Risk Manag. 2014, 10, 75–87. [Google Scholar]
- Hébert, M.; Lesept, F.; Vivien, D.; Macrez, R. The Story of an Exceptional Serine Protease, Tissue-Type Plasminogen Activator (TPA). Rev. Neurol. 2016, 172, 186–197. [Google Scholar] [CrossRef]
- Collen, D.; Lijnen, H.R. Tissue-Type Plasminogen Activator: A Historical Perspective and Personal Account. J. Thromb. Haemost. 2004, 2, 541–546. [Google Scholar] [CrossRef]
- Kwiatkowski, T.G.; Libman, R.B.; Frankel, M.; Tilley, B.C.; Morgenstern, L.B.; Lu, M.; Broderick, J.P.; Lewandowski, C.A.; Marler, J.R.; Levine, S.R.; et al. Effects of Tissue Plasminogen Activator for Acute Ischemic Stroke at One Year. National Institute of Neurological Disorders and Stroke Recombinant Tissue Plasminogen Activator Stroke Study Group. N. Engl. J. Med. 1999, 340, 1781–1787. [Google Scholar] [CrossRef]
- Schwammenthal, Y.; Drescher, M.J.; Merzeliak, O.; Tsabari, R.; Bruk, B.; Feibel, M.; Hoffman, C.; Bakon, M.; Rotstein, Z.; Chapman, J.; et al. Intravenous Recombinant Tissue Plasminogen Activator Therapy for Acute Ischemic Stroke: Initial Israeli Experience. Age 2004, 61, 69. [Google Scholar]
- Wardlaw, J.M.; Murray, V.; Berge, E.; del Zoppo, G.J. Thrombolysis for Acute Ischaemic Stroke. Cochrane Database Syst. Rev. 2014, 2014, CD000213. [Google Scholar] [CrossRef]
- Pennica, D.; Holmes, W.E.; Kohr, W.J.; Harkins, R.N.; Vehar, G.A.; Ward, C.A.; Bennett, W.F.; Yelverton, E.; Seeburg, P.H.; Heyneker, H.L.; et al. Cloning and Expression of Human Tissue-Type Plasminogen Activator CDNA in E. coli. Nature 1983, 301, 214–221. [Google Scholar] [CrossRef]
- Global Use of Strategies to Open Occluded Coronary Arteries (GUSTO III) Investigators. A Comparison of Reteplase with Alteplase for Acute Myocardial Infarction. N. Engl. J. Med. 1997, 337, 1118–1123. [Google Scholar] [CrossRef]
- Van de Werf, F.; Adgey, J.; Ardissino, D.; Armstrong, P.W.; Aylward, P.; Barbash, G.; Betriu, A.; Binbrek, A.S.; Califf, R.; Diaz, R.; et al. Single-Bolus Tenecteplase Compared with Front-Loaded Alteplase in Acute Myocardial Infarction: The ASSENT-2 Double-Blind Randomised Trial. Lancet 1999, 354, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Al-Nuaimi, Y.; Wainwright, J.; Sherrington, C.; Singh, A.; Kallingal, J.; Douglass, C.; Parry-Jones, A.; Smith, C.; Dixit, A.; et al. The Influence of Bolus to Infusion Delays on Plasma Tissue Plasminogen Activator Levels. Int. J. Stroke 2012, 9, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Nordt, T.K.; Bode, C. Thrombolysis: Newer Thrombolytic Agents and Their Role in Clinical Medicine. Heart 2003, 89, 1358–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potla, N.; Ganti, L. Tenecteplase vs. Alteplase for Acute Ischemic Stroke: A Systematic Review. Int. J. Emerg. Med. 2022, 15, 1–6. [Google Scholar] [CrossRef]
- Broderick, J.P. Intracerebral Hemorrhage After Intravenous T-PA Therapy for Ischemic Stroke. Stroke 1997, 28, 2109–2118. [Google Scholar]
- Man, S.; Xian, Y.; Holmes, D.N.; Matsouaka, R.A.; Saver, J.L.; Smith, E.E.; Bhatt, D.L.; Schwamm, L.H.; Fonarow, G.C. Association between Thrombolytic Door-to-Needle Time and 1-Year Mortality and Readmission in Patients with Acute Ischemic Stroke. JAMA J. Am. Med. Assoc. 2020, 323, 2170–2184. [Google Scholar] [CrossRef]
- Knecht, T.; Borlongan, C.; Peña, I. Combination Therapy for Ischemic Stroke: Novel Approaches to Lengthen Therapeutic Window of Tissue Plasminogen Activator. Brain Circ. 2018, 4, 99–108. [Google Scholar] [PubMed]
- Yang, S.H.; Liu, R. Four Decades of Ischemic Penumbra and Its Implication for Ischemic Stroke. Transl. Stroke Res. 2021, 12, 937–945. [Google Scholar] [CrossRef]
- Wu, J.; Echeverry, R.; Guzman, J.; Yepes, M. Neuroserpin Protects Neurons from Ischemia-Induced Plasmin-Mediated Cell Death Independently of Tissue-Type Plasminogen Activator Inhibition. Am. J. Pathol. 2010, 177, 2576–2584. [Google Scholar] [CrossRef]
- Xue, M.; Del Bigio, M.R. Acute Tissue Damage after Injections of Thrombin and Plasmin into Rat Striatum. Stroke 2001, 32, 2164–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Berthiller, J.; Derex, L.; Trouillas, P.; Diallo, L.; Hanss, M. Post-Thrombolysis Haemostasis Changes after Rt-PA Treatment in Acute Cerebral Infarct. Correlations with Cardioembolic Aetiology and Outcome. J. Neurol. Sci. 2015, 349, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.J.; Wang, S.X.; Chai, L.J.; Zhang, Y.; Guo, H.; Hu, L.M. Xueshuantong Injection (Lyophilized) Combined with Salvianolate Lyophilized Injection Protects against Focal Cerebral Ischemia/Reperfusion Injury in Rats through Attenuation of Oxidative Stress. Acta Pharmacol. Sin. 2018, 39, 998–1011. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.C.; Kong, Y.Y.; Hua, X.; Li, G.Q.; Zheng, S.L.; Cheng, M.H.; Wang, P.; Miao, C.Y. NAD Replenishment with Nicotinamide Mononucleotide Protects Blood-Brain Barrier Integrity and Attenuates Delayed Tissue Plasminogen Activator-Induced Haemorrhagic Transformation after Cerebral Ischaemia. Br. J. Pharmacol. 2017, 174, 3823–3836. [Google Scholar] [CrossRef] [Green Version]
- Kurzepa, J.; Kurzepa, J.; Golab, P.; Czerska, S.; Bielewicz, J. The Significance of Matrix Metalloproteinase (MMP)-2 and MMP-9 in the Ischemic Stroke. Int. J. Neurosci. 2014, 124, 707–716. [Google Scholar] [CrossRef]
- Leira, R.; Sobrino, T.; Blanco, M.; Campos, F.; Rodríguez-Ýñez, M.; Castellanos, M.; Moldes, O.; Millán, M.; Dávalos, A.; Castillo, J. A Higher Body Temperature Is Associated with Haemorrhagic Transformation in Patients with Acute Stroke Untreated with Recombinant Tissue-Type Plasminogen Activator (RtPA). Clin. Sci. 2012, 122, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Álvarez-Sabín, J.; Maisterra, O.; Santamarina, E.; Kase, C.S. Factors Influencing Haemorrhagic Transformation in Ischaemic Stroke. Lancet Neurol. 2013, 12, 689–705. [Google Scholar] [CrossRef]
- Montaner, J.; Ramiro, L.; Simats, A.; Hernández-Guillamon, M.; Delgado, P.; Bustamante, A.; Rosell, A. Matrix Metalloproteinases and ADAMs in Stroke. Cell. Mol. Life Sci. 2019, 76, 3117–3140. [Google Scholar] [CrossRef]
- Cheon, S.Y.; Kim, S.Y.; Kam, E.H.; Lee, J.H.; Kim, J.M.; Kim, E.J.; Kim, T.W.; Koo, B.N. Isoflurane Preconditioning Inhibits the Effects of Tissue-Type Plasminogen Activator on Brain Endothelial Cell in an in Vitro Model of Ischemic Stroke. Int. J. Med. Sci. 2017, 14, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Jin, X.; Liu, K.J.; Liu, W. Matrix Metalloproteinase-2-Mediated Occludin Degradation and Caveolin-1-Mediated Claudin-5 Redistribution Contribute to Blood-Brain Barrier Damage in Early Ischemic Stroke Stage. J. Neurosci. 2012, 32, 3044–3057. [Google Scholar] [CrossRef] [Green Version]
- Neuhaus, W.; Gaiser, F.; Mahringer, A.; Franz, J.; Riethmüller, C.; Förster, C. The Pivotal Role of Astrocytes in an in Vitro Stroke Model of the Blood-Brain Barrier. Front. Cell. Neurosci. 2014, 8, 352. [Google Scholar] [CrossRef] [Green Version]
- Kenna, J.E.; Anderton, R.S.; Knuckey, N.W.; Meloni, B.P. Assessment of Recombinant Tissue Plasminogen Activator (RtPA) Toxicity in Cultured Neural Cells and Subsequent Treatment with Poly-Arginine Peptide R18D. Neurochem. Res. 2020, 45, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Stanton, H.; Cowell, S.; Butler, G.; Knäuper, V.; Atkinson, S.; Gavrilovic, J. Mechanisms for pro Matrix Metalloproteinase Activation. APMIS 1999, 107, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Kovalevich, J.; Langford, D. Considerations for the Use of SH-SY5Y Neuroblastoma Cells in Neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar]
- Victoria, E.C.G.; de Brito Toscano, E.C.; Oliveira, F.M.S.; de Carvalho, B.A.; Caliari, M.V.; Teixeira, A.L.; de Miranda, A.S.; Rachid, M.A. Up-Regulation of Brain Cytokines and Metalloproteinases 1 and 2 Contributes to Neurological Deficit and Brain Damage in Transient Ischemic Stroke. Microvasc. Res. 2020, 129, 103973. [Google Scholar] [CrossRef]
- Ma, F.; Martínez-San Segundo, P.; Barceló, V.; Morancho, A.; Gabriel-Salazar, M.; Giralt, D.; Montaner, J.; Rosell, A. Matrix Metalloproteinase-13 Participates in Neuroprotection and Neurorepair after Cerebral Ischemia in Mice. Neurobiol. Dis. 2016, 91, 236–246. [Google Scholar] [CrossRef]
- Rodríguez, J.A.; Sobrino, T.; Orbe, J.; Purroy, A.; Martínez-Vila, E.; Castillo, J.; Páramo, J.A. ProMetalloproteinase-10 Is Associated with Brain Damage and Clinical Outcome in Acute Ischemic Stroke. J. Thromb. Haemost. 2013, 11, 1464–1473. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, E.; Rosell, A.; Penalba, A.; Slevin, M.; Alvarez-Sabín, J.; Ortega-Aznar, A.; Montaner, J. Vascular MMP-9/TIMP-2 and Neuronal MMP-10 up-Regulation in Human Brain after Stroke: A Combined Laser Microdissection and Protein Array Study. J. Proteome Res. 2009, 8, 3191–3197. [Google Scholar] [CrossRef] [PubMed]
- Palm, F.; Pussinen, P.J.; Safer, A.; Tervahartiala, T.; Sorsa, T.; Urbanek, C.; Becher, H.; Grau, A.J. Serum Matrix Metalloproteinase-8, Tissue Inhibitor of Metalloproteinase and Myeloperoxidase in Ischemic Stroke. Atherosclerosis 2018, 271, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challa, S.R.; Nalamolu, K.R.; Fornal, C.A.; Wang, B.C.; Martin, R.C.; Olson, E.A.; Ujjainwala, A.L.; Pinson, D.M.; Klopfenstein, J.D.; Veeravalli, K.K. Therapeutic Efficacy of Matrix Metalloproteinase-12 Suppression on Neurological Recovery after Ischemic Stroke: Optimal Treatment Timing and Duration. Front. Neurosci. 2022, 16, 1012812. [Google Scholar] [CrossRef]
- Arruri, V.; Chokkalla, A.K.; Jeong, S.; Chelluboina, B.; Mehta, S.L.; Veeravalli, K.K.; Vemuganti, R. MMP-12 Knockdown Prevents Secondary Brain Damage after Ischemic Stroke in Mice. Neurochem. Int. 2022, 161, 105432. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Nagai, N.; Umemura, K.; Collen, D.; Lijnen, H.R. Stromelysin-1 (MMP-3) Is Critical for Intracranial Bleeding after t-PA Treatment of Stroke in Mice. J. Thromb. Haemost. 2007, 5, 1732–1739. [Google Scholar] [CrossRef] [PubMed]
- Hohjoh, H.; Horikawa, I.; Nakagawa, K.; Segi-Nishida, E.; Hasegawa, H. Induced MRNA Expression of Matrix Metalloproteinases Mmp-3, Mmp-12, and Mmp-13 in the Infarct Cerebral Cortex of Photothrombosis Model Mice. Neurosci. Lett. 2020, 739, 135406. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.W.; Krekoski, C.A.; Bou, S.S.; Chapman, K.R.; Edwards, D.R. Increased Gelatinase A (MMP-2) and Gelatinase B (MMP-9) Activities in Human Brain after Focal Ischemia. Neurosci. Lett. 1997, 238, 53–56. [Google Scholar] [CrossRef]
- Planas, A.M.; Solé, S.; Justicia, C. Expression and Activation of Matrix Metalloproteinase-2 and -9 in Rat Brain after Transient Focal Cerebral Ischemia. Neurobiol. Dis. 2001, 8, 834–846. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.Q.; Wang, S.; Kim, H.Y.; Storrie, H.; Rosen, B.R.; Mooney, D.J.; Wang, X.; Lo, E.H. Role of Matrix Metalloproteinases in Delayed Cortical Responses after Stroke. Nat. Med. 2006, 12, 441–445. [Google Scholar] [CrossRef]
- Lapchak, P.A.; Chapman, D.F.; Zivin, J.A. Metalloproteinase Inhibition Reduces Thrombolytic (Tissue Plasminogen Activator)-Induced Hemorrhage after Thromboembolic Stroke. Stroke 2000, 31, 3034–3040. [Google Scholar] [CrossRef] [Green Version]
- Pfefferkorn, T.; Rosenberg, G.A. Closure of the Blood-Brain Barrier by Matrix Metalloproteinase Inhibition Reduces RtPA-Mediated Mortality in Cerebral Ischemia with Delayed Reperfusion. Stroke 2003, 34, 2025–2030. [Google Scholar] [CrossRef] [Green Version]
- Sumii, T.; Lo, E.H. Involvement of Matrix Metalloproteinase in Thrombolysis-Associated Hemorrhagic Transformation after Embolic Focal Ischemia in Rats. Stroke 2002, 33, 831–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lee, S.R.; Arai, K.; Lee, S.R.; Tsuji, K.; Rebeck, G.W.; Lo, E.H. Lipoprotein Receptor-Mediated Induction of Matrix Metalloproteinase by Tissue Plasminogen Activator. Nat. Med. 2003, 9, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Yang, J.; Tanaka, S.; Gonias, S.L.; Mars, W.M.; Liu, Y. Tissue-Type Plasminogen Activator Acts as a Cytokine That Triggers Intracellular Signal Transduction and Induces Matrix Metalloproteinase-9 Gene Expression. J. Biol. Chem. 2006, 281, 2120–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Nagai, N.; Yamakawa, K.; Kawakami, J.; Lijnen, H.R.; Umemura, K. Tissue-Type Plasminogen Activator (t-PA) Induces Stromelysin-1 (MMP-3) in Endothelial Cells through Activation of Lipoprotein Receptor-Related Protein. Blood 2009, 114, 3352–3358. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Cheng, Y.; Bi, G.; Zhu, Y.; Jun, W.; Ma, W.; Wu, H. Release of Matrix Metalloproteinases-2 and 9 by S-Nitrosylated Caveolin-1 Contributes to Degradation of Extracellular Matrix in TPA-Treated Hypoxic Endothelial Cells. PLoS ONE 2016, 11, e0149269. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; An, J.; Haile, W.B.; Echeverry, R.; Strickland, D.K.; Yepes, M. Microglial Low-Density Lipoprotein Receptor-Related Protein 1 Mediates the Effect of Tissue-Type Plasminogen Activator on Matrix Metalloproteinase-9 Activity in the Ischemic Brain. J. Cereb. Blood Flow Metab. 2009, 29, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Siniatchkin, M.; Averkina, N.; Andrasik, F.; Stephani, U.; Gerber, W.D. Neurophysiological Reactivity before a Migraine Attack. Neurosci. Lett. 2006, 400, 121–124. [Google Scholar] [CrossRef]
- Turner, R.J.; Sharp, F.R. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front. Cell. Neurosci. 2016, 10, 56. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, E.; Ortega, L.; Hernández-Guillamon, M.; Penalba, A.; Fernández-Cadenas, I.; Rosell, A.; Montaner, J. Tissue Plasminogen Activator (t-PA) Promotes Neutrophil Degranulation and MMP-9 Release. J. Leukoc. Biol. 2008, 84, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Gautier, S.; Ouk, T.; Tagzirt, M.; Lefebvre, C.; Laprais, M.; Pétrault, O.; Dupont, A.; Leys, D.; Bordet, R. Impact of the Neutrophil Response to Granulocyte Colony-Stimulating Factor on the Risk of Hemorrhage When Used in Combination with Tissue Plasminogen Activator during the Acute Phase of Experimental Stroke. J. Neuroinflamm. 2014, 11, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.W.; Wang, M.J.; Gong, S.S.; Sun, M.H.; Wang, Y.; Zhang, Y.Y.; Li, F.; Yu, B.Y.; Kou, J.P. YiQiFuMai Lyophilized Injection Ameliorates TPA-Induced Hemorrhagic Transformation by Inhibiting Cytoskeletal Rearrangement Associated with ROCK1 and NF-ΚB Signaling Pathways. J. Ethnopharmacol. 2020, 262, 113161. [Google Scholar] [CrossRef] [PubMed]
- Gerzanich, V.; Kwon, M.S.; Woo, S.K.; Ivanov, A.; Marc Simard, J. SUR1-TRPM4 Channel Activation and Phasic Secretion of MMP-9 Induced by TPA in Brain Endothelial Cells. PLoS ONE 2018, 13, e0195526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, S.; Lee, J.H.; Wali, B.; Stein, D.G.; Sayeed, I. Progesterone Attenuates Hemorrhagic Transformation after Delayed TPA Treatment in an Experimental Model of Stroke in Rats: Involvement of the VEGF–MMP Pathway. J. Cereb. Blood Flow Metab. 2014, 34, 72. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Fan, W.; Cai, P.; Fan, M.; Zhu, X.; Dai, Y.; Sun, C.; Cheng, Y.; Zheng, P.; Zhao, B.Q. Recombinant ADAMTS13 Reduces Tissue Plasminogen Activator-Induced Hemorrhage after Stroke in Mice. Ann. Neurol. 2013, 73, 189–198. [Google Scholar] [CrossRef]
- Narasimhan, P.; Liu, J.; Song, Y.S.; Massengale, J.L.; Chan, P.H. VEGF Stimulates the ERK 1/2 Signaling Pathway and Apoptosis in Cerebral Endothelial Cells After Ischemic Conditions. Stroke J. Cereb. Circ. 2009, 40, 1467. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.Q.; Li, W.; Li, S.Q.; Li, J.; Li, Y.W.; Kong, S.X.; Liu, R.M.; Wang, S.M.; Lv, W.M. MCP-1 Stimulates MMP-9 Expression via ERK 1/2 and P38 MAPK Signaling Pathways in Human Aortic Smooth Muscle Cells. Cell. Physiol. Biochem. 2014, 34, 266–276. [Google Scholar] [CrossRef]
- Yu, S.M.; Kim, S.J. Salinomycin Causes Migration and Invasion of Human Fibrosarcoma Cells by Inducing MMP-2 Expression via PI3-Kinase, ERK-1/2 and P38 Kinase Pathways. Int. J. Oncol. 2016, 48, 2686–2692. [Google Scholar] [CrossRef] [Green Version]
- Ismael, S.; Nasoohi, S.; Yoo, A.; Ahmed, H.A.; Ishrat, T. Tissue Plasminogen Activator Promotes TXNIP-NLRP3 Inflammasome Activation after Hyperglycemic Stroke in Mice. Mol. Neurobiol. 2020, 57, 2495–2508. [Google Scholar] [CrossRef]
- Tang, G.; Liu, Y.; Zhang, Z.; Lu, Y.; Wang, Y.; Huang, J.; Li, Y.; Chen, X.; Gu, X.; Wang, Y.; et al. Mesenchymal Stem Cells Maintain Blood-Brain Barrier Integrity by Inhibiting Aquaporin-4 Upregulation after Cerebral Ischemia. Stem Cells 2014, 32, 3150–3162. [Google Scholar] [CrossRef]
- Anfray, A.; Drieu, A.; Hingot, V.; Hommet, Y.; Yetim, M.; Rubio, M.; Deffieux, T.; Tanter, M.; Orset, C.; Vivien, D. Circulating TPA Contributes to Neurovascular Coupling by a Mechanism Involving the Endothelial NMDA Receptors. J. Cereb. Blood Flow Metab. 2020, 40, 2038–2054. [Google Scholar] [CrossRef] [PubMed]
- Correa, F.; Gauberti, M.; Parcq, J.; Macrez, R.; Hommet, Y.; Obiang, P.; Hernangómez, M.; Montagne, A.; Liot, G.; Guaza, C.; et al. Tissue Plasminogen Activator Prevents White Matter Damage Following Stroke. J. Exp. Med. 2011, 208, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, J.; Otter, M.; Rijken, D.C.; Van Berkel, T.J.C. Characterization of the Interaction in Vivo of Tissue-Type Plasminogen Activator with Liver Cells. J. Biol. Chem. 1988, 263, 18220–18224. [Google Scholar] [CrossRef] [PubMed]
- White, S.; Lin, L.; Hu, K. NF-ΚB and TPA Signaling in Kidney and Other Diseases. Cells 2020, 9, 1348. [Google Scholar] [CrossRef] [PubMed]
- Etique, N.; Verzeaux, L.; Dedieu, S.; Emonard, H. Lrp-1: A Checkpoint for the Extracellular Matrix Proteolysis. BioMed Res. Int. 2013, 2013, 152163. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Cheon, S.Y.; Kim, E.J.; Lee, J.H.; Kam, E.H.; Kim, J.M.; Park, M.; Koo, B.N. Isoflurane Postconditioning Inhibits TPA-Induced Matrix Metalloproteinases Activation After Hypoxic Injury via Low-Density Lipoprotein Receptor-Related Protein and Extracellular Signal-Regulated Kinase Pathway. Neurochem. Res. 2017, 21, 160–174. [Google Scholar] [CrossRef]
- Lapchak, P.A.; Han, M.K. Simvastatin Improves Clinical Scores in a Rabbit Multiple Infarct Ischemic Stroke Model: Synergism with a ROCK Inhibitor but Not the Thrombolytic Tissue Plasminogen Activator. Brain Res. 2010, 1344, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Elshiaty, M.; Schindler, H.; Christopoulos, P. Principles and Current Clinical Landscape of Multispecific Antibodies against Cancer. Int. J. Mol. Sci. 2021, 22, 5632. [Google Scholar] [CrossRef]
- Rikitake, Y.; Kim, H.H.; Huang, Z.; Seto, M.; Yano, K.; Asano, T.; Moskowitz, M.A.; Liao, J.K. Inhibition of Rho Kinase (ROCK) Leads to Increased Cerebral Blood Flow and Stroke Protection. Stroke 2005, 36, 2251–2257. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.A.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Simvastatin Inhibits MMP-9 Secretion from Human Saphenous Vein Smooth Muscle Cells by Inhibiting the RhoA/ROCK Pathway and Reducing MMP-9 MRNA Levels. FASEB J. 2005, 19, 804–806. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S. TPA Helpers in the Treatment of Acute Ischemic Stroke: Are They Ready for Clinical Use? J. Stroke 2019, 21, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Paemen, L.; Martens, E.; Norga, K.; Masure, S.; Roets, E.; Hoogmartens, J.; Opdenakker, G. The Gelatinase Inhibitory Activity of Tetracyclines and Chemically Modified Tetracycline Analogues as Measured by a Novel Microtiter Assay for Inhibitors. Biochem. Pharmacol. 1996, 52, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Golub, L.M.; Ramamurthy, N.; McNamara, T.F.; Gomes, B.; Wolff, M.; Casino, A.; Kapoor, A.; Zambon, J.; Ciancio, S.; Schneir, M.; et al. Tetracyclines Inhibit Tissue Collagenase Activity. A New Mechanism in the Treatment of Periodontal Disease. J. Periodontal Res. 1984, 19, 651–655. [Google Scholar] [CrossRef]
- Murata, Y.; Rosell, A.; Scannevin, R.H.; Rhodes, K.J.; Wang, X.; Lo, E.H. Extension of the Thrombolytic Time Window with Minocycline in Experimental Stroke. Stroke 2008, 39, 3372–3377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagan, S.C.; Waller, J.L.; Nichols, F.T.; Edwards, D.J.; Pettigrew, L.C.; Clark, W.M.; Hall, C.E.; Switzer, J.A.; Ergul, A.; Hess, D.C. Minocycline to Improve Neurologic Outcome in Stroke (MINOS): A Dose-Finding Study. Stroke 2010, 41, 2283–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, S.J.; Lee, S.H.; Shin, K.Y.; Lee, C.K.; Cho, I.H.; Kim, H.S.; Suh, Y.H. SP-8203 Reduces Oxidative Stress via SOD Activity and Behavioral Deficit in Cerebral Ischemia. Pharmacol. Biochem. Behav. 2011, 98, 150–154. [Google Scholar] [CrossRef]
- Noh, S.J.; Lee, J.M.; Lee, K.S.; Hong, H.S.; Lee, C.K.; Cho, I.H.; Kim, H.S.; Suh, Y.H. SP-8203 Shows Neuroprotective Effects and Improves Cognitive Impairment in Ischemic Brain Injury through NMDA Receptor. Pharmacol. Biochem. Behav. 2011, 100, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Song, H.Y.; Chung, J.I.; Anthony Jalin, A.M.; Ju, C.; Pahk, K.; Joung, C.; Lee, S.; Jin, S.; Kim, B.S.; Lee, K.S.; et al. The Quinazoline Otaplimastat (SP-8203) Reduces the Hemorrhagic Transformation and Mortality Aggravated after Delayed RtPA-Induced Thrombolysis in Cerebral Ischemia. Int. J. Mol. Sci. 2022, 23, 1403. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, K.B.; Park, J.H.; Sung, S.M.; Oh, K.; Kim, E.G.; Chang, D.I.; Hwang, Y.H.; Lee, E.J.; Kim, W.K.; et al. Safety and Efficacy of Otaplimastat in Patients with Acute Ischemic Stroke Requiring TPA (SAFE-TPA): A Multicenter, Randomized, Double-Blind, Placebo-Controlled Phase 2 Study. Ann. Neurol. 2020, 87, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Pervin, M.; Unno, K.; Takagaki, A.; Isemura, M.; Nakamura, Y. Function of Green Tea Catechins in the Brain: Epigallocatechin Gallate and Its Metabolites. Int. J. Mol. Sci. 2019, 20, 3630. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Coria, H.; Arrieta-Cruz, I.; Gutiérrez-Juárez, R.; López-Valdés, H.E. Anti-Inflammatory Effects of Flavonoids in Common Neurological Disorders Associated with Aging. Int. J. Mol. Sci. 2023, 24, 4297. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Wang, M.; Jing, X.; Shi, H.; Ren, M.; Lou, H. (-)-Epigallocatechin Gallate Protects against Cerebral Ischemia-Induced Oxidative Stress via Nrf2/ARE Signaling. Neurochem. Res. 2014, 39, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Kim, H.S.; Kim, Y.K.; Kim, T.M.; Im, S.; Chung, M.E.; Hong, B.Y.; Ko, Y.J.; Kim, H.W.; Lee, J.I. The Functional Effect of Epigallocatechin Gallate on Ischemic Stroke in Rats. Acta Neurobiol. Exp. (Wars) 2010, 70, 40–46. [Google Scholar]
- You, Y.P. Epigallocatechin Gallate Extends the Therapeutic Window of Recombinant Tissue Plasminogen Activator Treatment in Ischemic Rats. J. Stroke Cerebrovasc. Dis. 2016, 25, 990–997. [Google Scholar] [CrossRef]
- Wang, X.H.; You, Y.P. Epigallocatechin Gallate Extends Therapeutic Window of Recombinant Tissue Plasminogen Activator Treatment for Brain Ischemic Stroke: A Randomized Double-Blind and Placebo-Controlled Trial. Clin. Neuropharmacol. 2017, 40, 24–28. [Google Scholar] [CrossRef]
- Borlongan, C.V.; Lind, J.G.; Dillon-Carter, O.; Yu, G.; Hadman, M.; Cheng, C.; Carroll, J.; Hess, D.C. Bone Marrow Grafts Restore Cerebral Blood Flow and Blood Brain Barrier in Stroke Rats. Brain Res. 2004, 1010, 108–116. [Google Scholar] [CrossRef]
- Park, H.J.; Shin, J.Y.; Kim, H.N.; Oh, S.H.; Song, S.K.; Lee, P.H. Mesenchymal Stem Cells Stabilize the Blood-Brain Barrier through Regulation of Astrocytes. Stem Cell Res. Ther. 2015, 6, 187. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Wang, L.; Qu, M.; Liang, H.; Li, W.; Li, Y.; Deng, L.; Zhang, Z.; Yang, G.Y. Mesenchymal Stem Cells Attenuate Blood-Brain Barrier Leakage after Cerebral Ischemia in Mice. J. Neuroinflamm. 2018, 15, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, P.T.; Wu, C.C.; Chiang, Y.H.; Hu, C.J.; Chen, K.Y. Mesenchymal Stem/Stromal Cell Therapy in Blood-Brain Barrier Preservation Following Ischemia: Molecular Mechanisms and Prospects. Int. J. Mol. Sci. 2021, 22, 10045. [Google Scholar] [CrossRef]
- Ding, X.; Li, Y.; Liu, Z.; Zhang, J.; Cui, Y.; Chen, X.; Chopp, M. The Sonic Hedgehog Pathway Mediates Brain Plasticity and Subsequent Functional Recovery after Bone Marrow Stromal Cell Treatment of Stroke in Mice. J. Cereb. Blood Flow Metab. 2013, 33, 1015–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, H.; Li, Y.; Shen, L.H.; Liu, X.; Hozeska-Solgot, A.; Zhang, R.L.; Zhang, Z.G.; Chopp, M. Multipotent Mesenchymal Stromal Cells Increase TPA Expression and Concomitantly Decrease PAI-1 Expression in Astrocytes through the Sonic Hedgehog Signaling Pathway after Stroke (in Vitro Study). J. Cereb. Blood Flow Metab. 2011, 31, 2181–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanotti, L.; Angioni, R.; Calì, B.; Soldani, C.; Ploia, C.; Moalli, F.; Gargesha, M.; D’Amico, G.; Elliman, S.; Tedeschi, G.; et al. Mouse Mesenchymal Stem Cells Inhibit High Endothelial Cell Activation and Lymphocyte Homing to Lymph Nodes by Releasing TIMP-1. Leukemia 2016, 30, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Ries, C.; Egea, V.; Karow, M.; Kolb, H.; Jochum, M.; Neth, P. MMP-2, MT1-MMP, and TIMP-2 Are Essential for the Invasive Capacity of Human Mesenchymal Stem Cells: Differential Regulation by Inflammatory Cytokines. Blood 2007, 109, 4055–4063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, T.; Gao, W.; Zhu, L.; Ren, J.; Yao, H.; Wang, K.; Shi, D. TIMP-1 Inhibits Proliferation and Osteogenic Differentiation of HBMSCs through Wnt/β-Catenin Signaling. Biosci. Rep. 2019, 39, BSR20181290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manji, H.K.; Lenox, R.H. Lithium: A Molecular Transducer of Mood-Stabilization in the Treatment of Bipolar Disorder. Neuropsychopharmacology 1998, 19, 161–166. [Google Scholar]
- Haupt, M.; Zechmeister, B.; Bosche, B.; Lieschke, S.; Zheng, X.; Zhang, L.; Venkataramani, V.; Jin, F.; Hein, K.; Weber, M.S.; et al. Lithium Enhances Post-Stroke Blood-Brain Barrier Integrity, Activates the MAPK/ERK1/2 Pathway and Alters Immune Cell Migration in Mice. Neuropharmacology 2020, 181, 108357. [Google Scholar] [CrossRef]
- Ji, Y.B.; Gao, Q.; Tan, X.X.; Huang, X.W.; Ma, Y.Z.; Fang, C.; Wang, S.N.; Qiu, L.H.; Cheng, Y.X.; Guo, F.Y.; et al. Lithium Alleviates Blood-Brain Barrier Breakdown after Cerebral Ischemia and Reperfusion by Upregulating Endothelial Wnt/β-Catenin Signaling in Mice. Neuropharmacology 2021, 186, 108474. [Google Scholar] [CrossRef]
- Silachev, D.N.; Gulyaev, M.V.; Zorova, L.D.; Khailova, L.S.; Gubsky, L.V.; Pirogov, Y.A.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Magnetic Resonance Spectroscopy of the Ischemic Brain under Lithium Treatment. Link to Mitochondrial Disorders under Stroke. Chem. Biol. Interact. 2015, 237, 175–182. [Google Scholar] [CrossRef]
- Silachev, D.N.; Plotnikov, E.Y.; Babenko, V.A.; Savchenko, E.S.; Zorova, L.D.; Pevzner, I.B.; Gulyaev, M.V.; Pirogov, Y.A.; Sukhikh, G.T.; Zorov, D.B. Protection of Neurovascular Unit Cells with Lithium Chloride and Sodium Valproate Prevents Brain Damage in Neonatal Ischemia/Hypoxia. Bull. Exp. Biol. Med. 2016, 160, 313–318. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babenko, V.A.; Fedulova, K.S.; Silachev, D.N.; Rahimi-Moghaddam, P.; Kalyuzhnaya, Y.N.; Demyanenko, S.V.; Plotnikov, E.Y. The Role of Matrix Metalloproteinases in Hemorrhagic Transformation in the Treatment of Stroke with Tissue Plasminogen Activator. J. Pers. Med. 2023, 13, 1175. https://doi.org/10.3390/jpm13071175
Babenko VA, Fedulova KS, Silachev DN, Rahimi-Moghaddam P, Kalyuzhnaya YN, Demyanenko SV, Plotnikov EY. The Role of Matrix Metalloproteinases in Hemorrhagic Transformation in the Treatment of Stroke with Tissue Plasminogen Activator. Journal of Personalized Medicine. 2023; 13(7):1175. https://doi.org/10.3390/jpm13071175
Chicago/Turabian StyleBabenko, Valentina A., Ksenia S. Fedulova, Denis N. Silachev, Parvaneh Rahimi-Moghaddam, Yulia N. Kalyuzhnaya, Svetlana V. Demyanenko, and Egor Y. Plotnikov. 2023. "The Role of Matrix Metalloproteinases in Hemorrhagic Transformation in the Treatment of Stroke with Tissue Plasminogen Activator" Journal of Personalized Medicine 13, no. 7: 1175. https://doi.org/10.3390/jpm13071175