
Higher pNRF2, SOCS3, IRF3, and RIG1 Tissue Protein Expression in NASH Patients versus NAFL Patients: pNRF2 Expression Is Concomitantly Associated with Elevated Fasting Glucose Levels
 , ,
, ,
Abstract
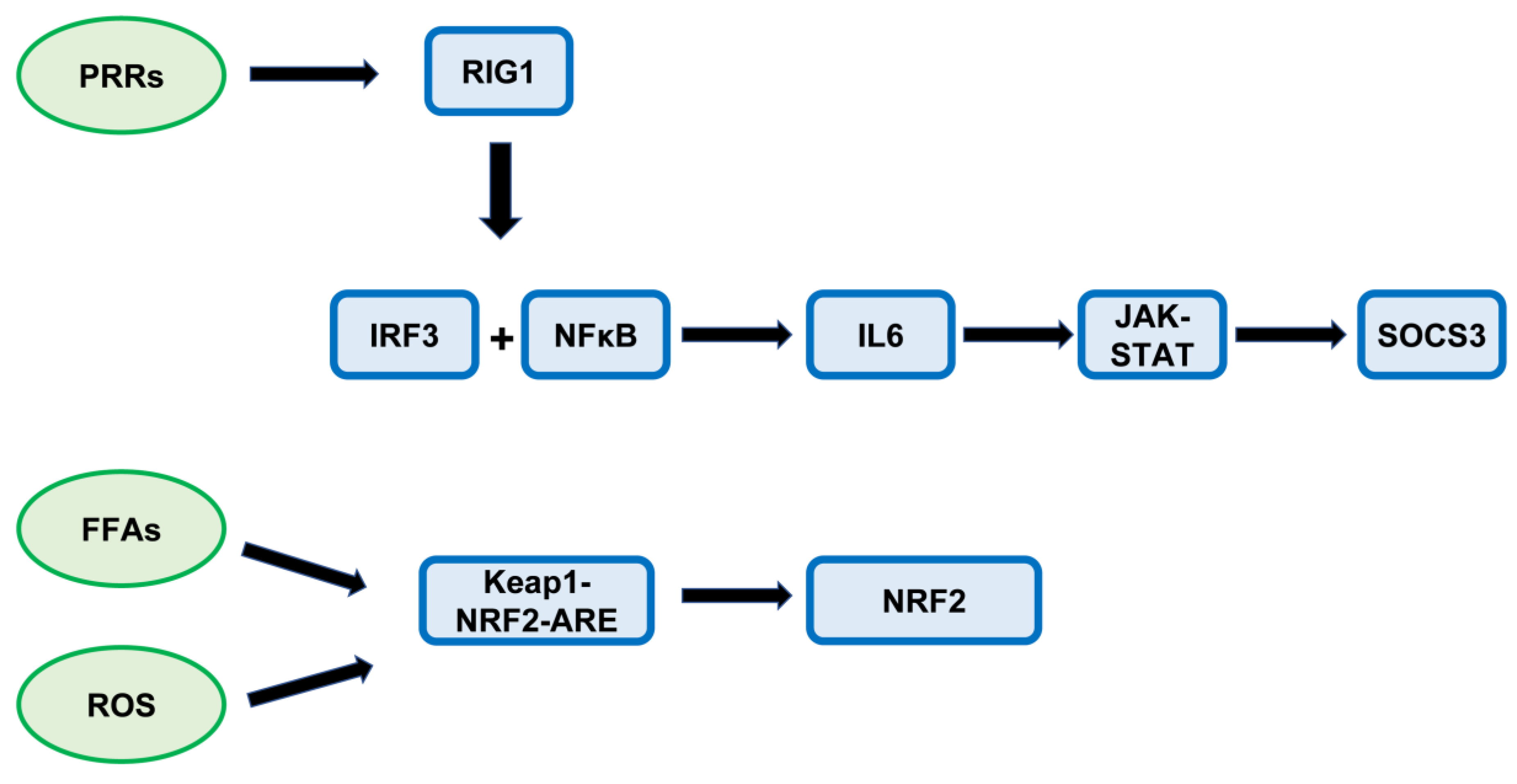
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Histology and Immunohistochemistry
2.3. Sample and Immunohistochemistry Evaluation
2.4. Statistics
3. Results
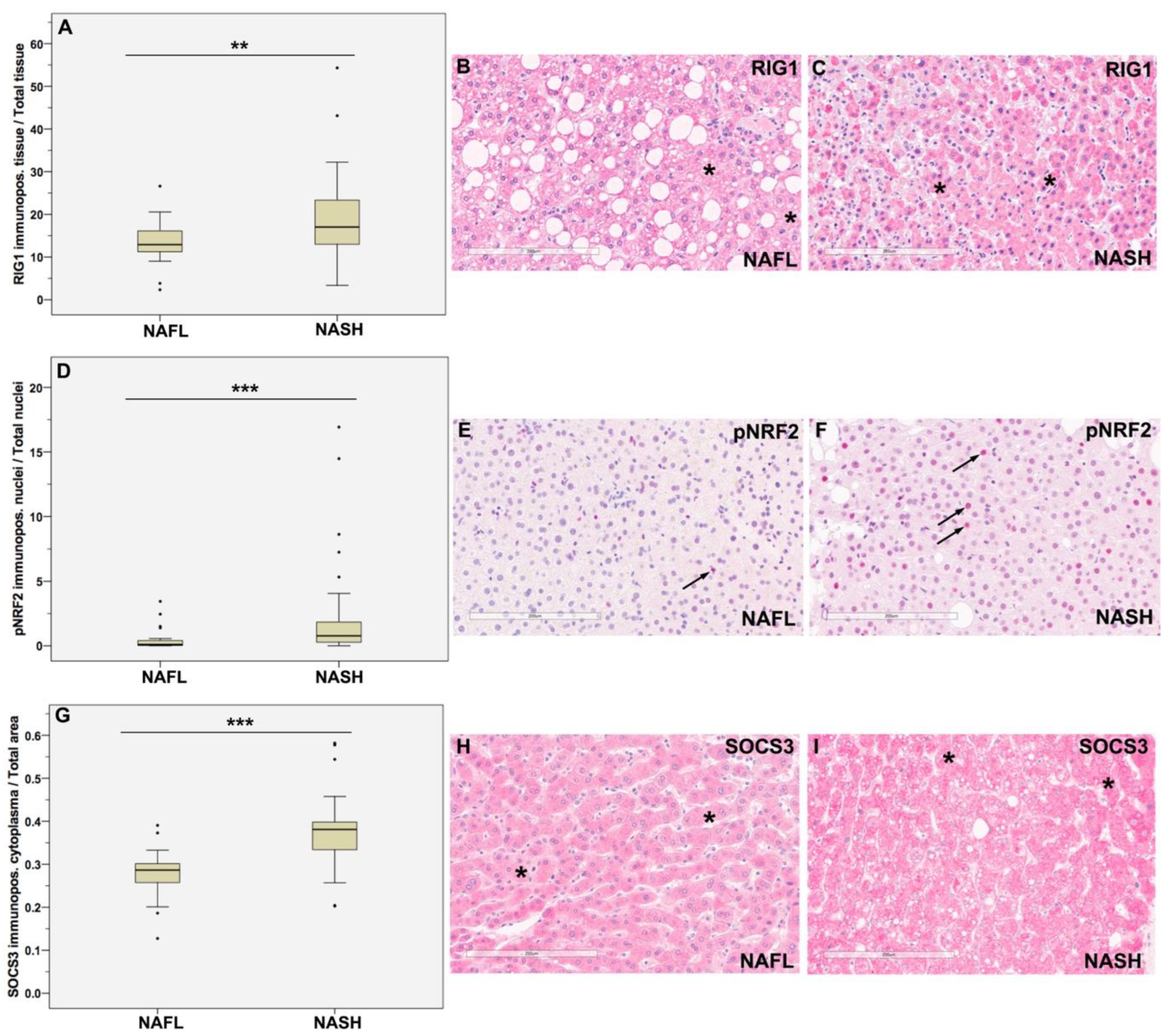
3.1. Higher Expression of RIG1, pNRF2, and SOCS3 in NASH vs. NAFL
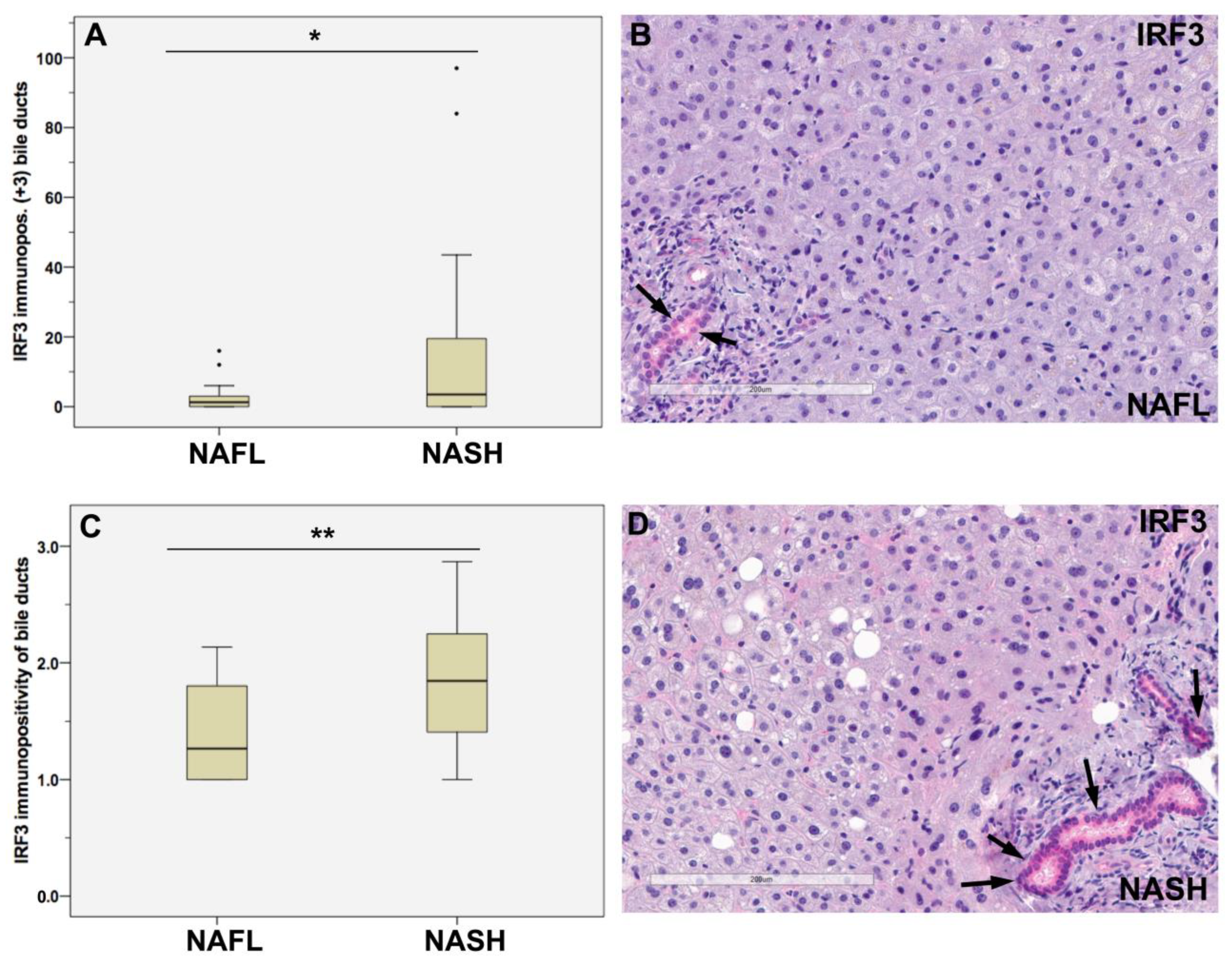
3.2. Stronger IRF3 Immunostaining of Bile Ducts in NASH Than in NAFL
3.3. Association between RIG1, pNRF2, SOCS3, IRF3 Immunopositivity and Histological/Laboratory Parameters
3.4. Multivariable Binary Logistic Regression Analysis
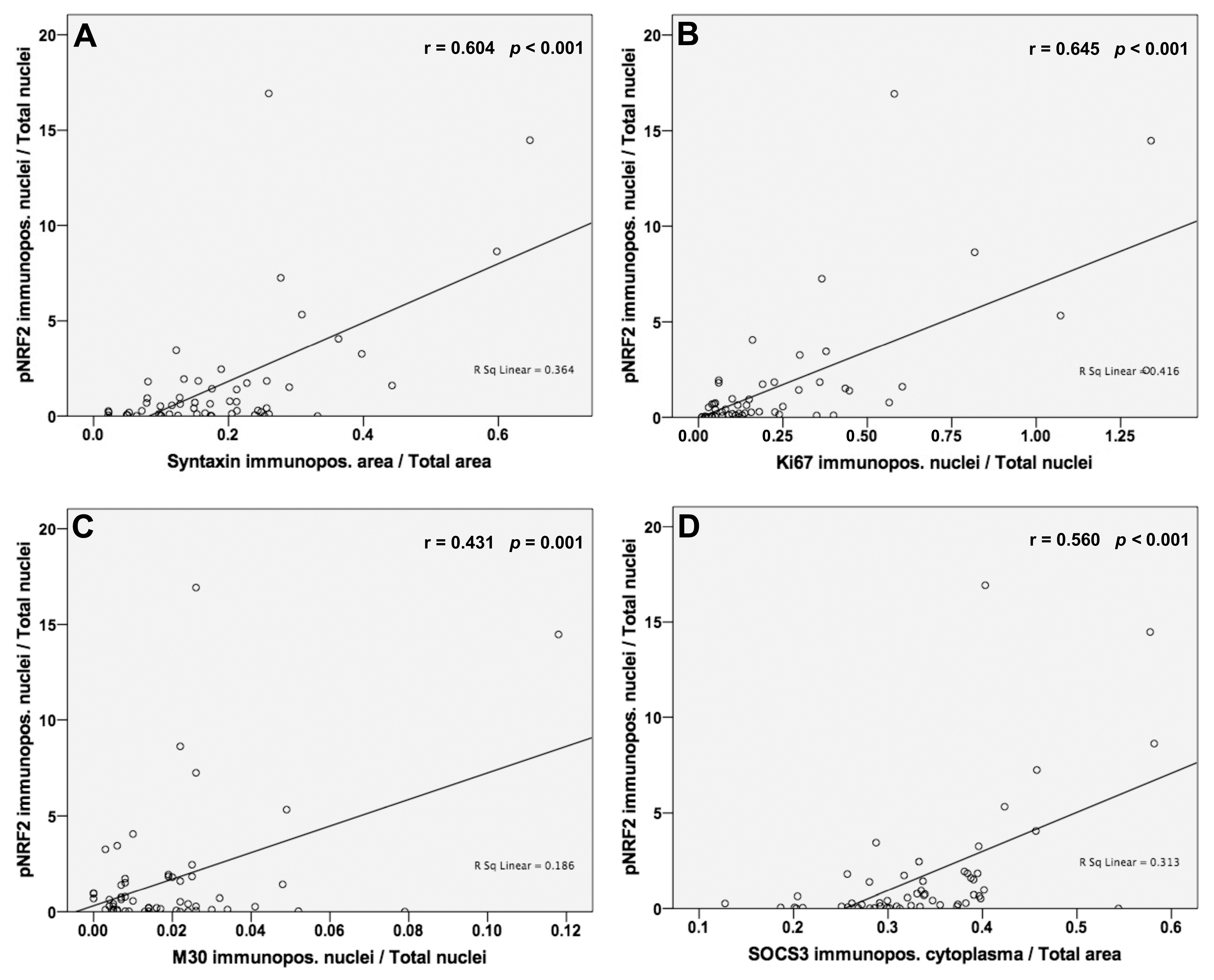
3.5. Analysis of Correlations between NRF2 Activation and Syntaxin, Ki67, M30 and SOCS3 Protein Levels
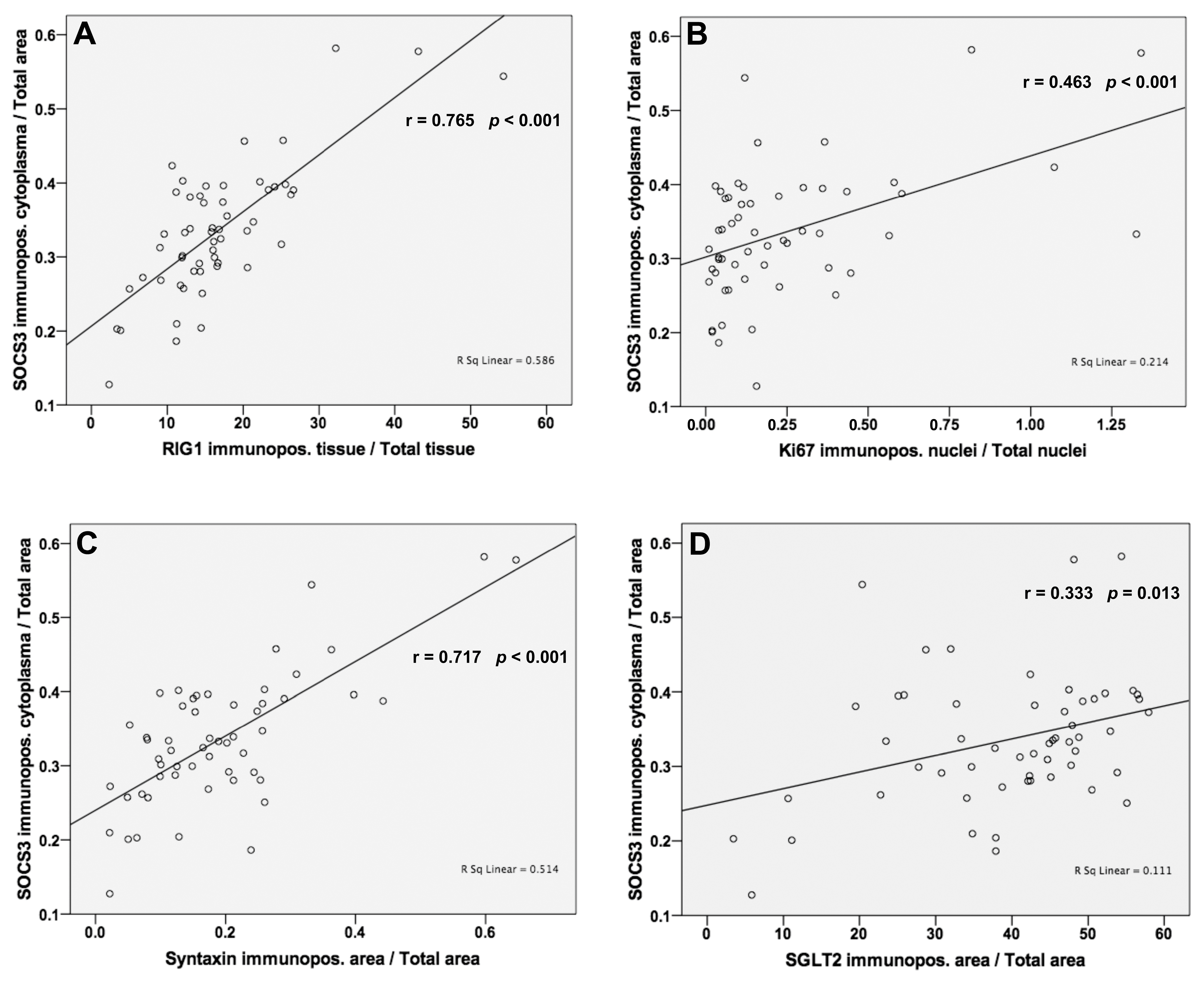
3.6. Analysis of Correlations between SOCS3 and RIG1, Ki67, Syntaxin and SGLT2 Protein Levels
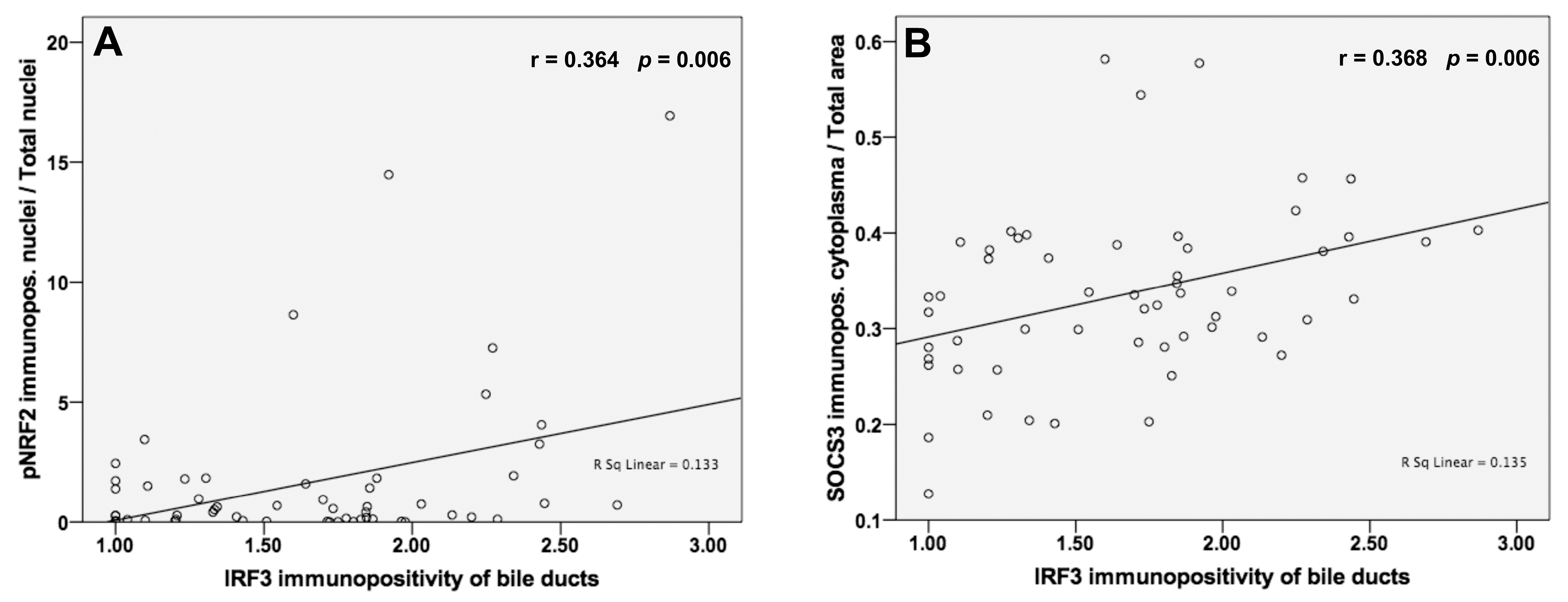
3.7. Analysis of Correlations between IRF3 and pNRF2 and SOCS3 Protein Levels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bai, L.; Li, H. Innate immune regulatory networks in hepatic lipid metabolism. J. Mol. Med. 2019, 97, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Cheung, S.T. STAT3: An emerging therapeutic target for hepatocellular carcinoma. Cancers 2019, 11, 1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naka, T.; Narazaki, M.; Hirata, M.; Matsumoto, T.; Minamoto, S.; Aono, A.; Nishimoto, N.; Kajita, T.; Taga, T.; Yoshizaki, K.; et al. Structure and function of a new STAT-induced STAT inhibitor. Nature 1997, 387, 924–929. [Google Scholar] [CrossRef]
- Jorgensen, S.B.; O’Neill, H.M.; Sylow, L.; Honeyman, J.; Hewitt, K.A.; Palanivel, R.; Fullerton, M.D.; Oberg, L.; Balendran, A.; Galic, S.; et al. Deletion of skeletal muscle SOCS3 prevents insulin resistance in obesity. Diabetes 2013, 62, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Chambel, S.S.; Santos-Goncalves, A.; Duarte, T.L. The dual role of Nrf2 in nonalcoholic fatty liver disease: Regulation of antioxidant defenses and hepatic lipid metabolism. BioMed Res. Int. 2015, 2015, 597134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Horst, D.; Carter-Timofte, M.E.; van Grevenynghe, J.; Laguette, N.; Dinkova-Kostova, A.T.; Olagnier, D. Regulation of innate immunity by Nrf2. Curr. Opin. Immunol. 2022, 78, 102247. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Koh, D.H.; Jun, D.W.; Roh, Y.J.; Kang, H.T.; Oh, J.H.; Kim, H. Auranofin attenuates hepatic steatosis and fibrosis in nonalcoholic fatty liver disease via NRF2 and NF-κB signaling pathways. Clin. Mol. Hepatol. 2022, 28, 827–840. [Google Scholar] [CrossRef]
- Bataille, A.M.; Manautou, J.E. Nrf2: A potential target for new therapeutics in liver disease. Clin. Pharmacol. Ther. 2012, 92, 340–348. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.K.K.; Zhang, L.; Chan, M.T.V. Autophagy, NAFLD and NAFLD-Related HCC. Obes. Fat. Liver Liver Cancer 2018, 1061, 127–138. [Google Scholar]
- Delgado, M.; Singh, S.; De Haro, S.; Master, S.; Ponpuak, M.; Dinkins, C.; Ornatowski, W.; Vergne, I.; Deretic, V. Autophagy and pattern recognition receptors in innate immunity. Immunol. Rev. 2009, 227, 189–202. [Google Scholar] [CrossRef]
- Wang, Y.; Singh, R.; Xiang, Y.; Czaja, M.J. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology 2010, 52, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Martinet, W.; Roth, L.; De Meyer, G.R.Y. Standard immunohistochemical assays to assess autophagy in mammalian tissue. Cells 2017, 6, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshii, S.R.; Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Qiang, S.; Nakatsu, Y.; Seno, Y.; Fujishiro, M.; Sakoda, H.; Kushiyama, A.; Mori, K.; Matsunaga, Y.; Yamamotoya, T.; Kamata, H.; et al. Treatment with the SGLT2 inhibitor luseogliflozin improves nonalcoholic steatohepatitis in a rodent model with diabetes mellitus. Diabetol. Metab. Syndr. 2015, 7, 104. [Google Scholar] [CrossRef] [Green Version]
- Jojima, T.; Wakamatsu, S.; Kase, M.; Iijima, T.; Maejima, Y.; Shimomura, K.; Kogai, T.; Tomaru, T.; Usui, I.; Aso, Y. The SGLT2 inhibitor canagliflozin prevents carcinogenesis in a mouse model of diabetes and non-alcoholic steatohepatitis-related hepatocarcinogenesis: Association with SGLT2 expression in hepatocellular carcinoma. Int. J. Mol. Sci. 2019, 20, 5237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, H.; Lee, J.-C.; Huang, H.-C.; Huang, H.; Liu, H.-K.; Huang, C. Delayed intervention with a novel SGLT2 inhibitor NGI001 suppresses diet-induced metabolic dysfunction and non-alcoholic fatty liver disease in mice. Br. J. Pharmacol. 2020, 177, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Hsiang, J.C.; Wong, V.W.-S. SGLT2 inhibitors in liver patients. Clin. Gastroenterol. Hepatol. 2020, 18, 2168–2172. [Google Scholar] [CrossRef] [PubMed]
- Kälsch, J.; Bechmann, L.P.; Kälsch, H.; Schlattjan, M.; Erhard, J.; Gerken, G.; Canbay, A. Evaluation of biomarkers of NAFLD in a cohort of morbidly obese patients. J. Nutr. Metab. 2011, 2011, 369168. [Google Scholar] [CrossRef] [Green Version]
- Ikura, Y. Transitions of histopathologic criteria for diagnosis of nonalcoholic fatty liver disease during the last three decades. World J. Hepatol. 2014, 6, 894–900. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P.; FLIP, P.C. Utility and appropriateness of the fatty liver inhibition of progression (FLIP) algorithm and steatosis, activity, and fibrosis (SAF) score in the evaluation of biopsies of nonalcoholic fatty liver disease. Hepatology 2014, 60, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Schwertheim, S.; Kälsch, J.; Jastrow, H.; Schaefer, C.M.; Theurer, S.; Ting, S.; Canbay, A.; Wedemeyer, H.; Schmid, K.W.; Baba, H.A. Characterization of two types of intranuclear hepatocellular inclusions in NAFLD. Sci. Rep. 2020, 10, 16533. [Google Scholar] [CrossRef]
- Remmele, W.; Stegner, H.E. Recommendation for uniform definition of an immunoreactive score (IRS) for immunohistochemical estrogen receptor detection (ER-ICA) in breast cancer tissue. Pathologe 1987, 8, 138–140. [Google Scholar]
- Bertram, S.; Padden, J.; Kälsch, J.; Ahrens, M.; Pott, L.; Canbay, A.; Weber, F.; Fingas, C.; Hoffmann, A.C.; Vietor, A.; et al. Novel immunohistochemical markers differentiate intrahepatic cholangiocarcinoma from benign bile duct lesions. J. Clin. Pathol. 2016, 69, 619–626. [Google Scholar] [CrossRef]
- Kälsch, J.; Padden, J.; Bertram, S.; Pott, L.L.; Reis, H.; Westerwick, D.; Schaefer, C.M.; Sowa, J.-P.; Möllmann, D.; Fingas, C.; et al. Annexin A10 optimally differentiates between intrahepatic cholangiocarcinoma and hepatic metastases of pancreatic ductal adenocarcinoma: A comparative study of immunohistochemical markers and panels. Virchows Arch. 2017, 470, 537–543. [Google Scholar] [CrossRef]
- Battifora, H. Assessment of antigen damage in immunohistochemistry: The vimentin internal control. Am. J. Clin. Pathol. 1991, 96, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Bertram, S.; Myland, C.; Swoboda, S.; Gallinat, A.; Minor, T.; Lehmann, N.; Thie, M.; Kälsch, J.; Pott, L.; Canbay, A.; et al. Liver steatosis in pre-transplant liver biopsies can be quantified rapidly and accurately by nuclear magnetic resonance analysis. Virchows Arch. 2017, 470, 197–204. [Google Scholar] [CrossRef]
- Apopa, P.L.; He, X.; Ma, Q. Phosphorylation of Nrf2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of Nrf2 in IMR-32 neuroblastoma cells. J. Biochem. Mol. Toxicol. 2008, 22, 63–76. [Google Scholar] [CrossRef]
- Huang, H.-C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Teng, D.; Shi, X.; Qin, G.; Qin, Y.; Quan, H.; Shi, B.; Sun, H.; Ba, J.; Chen, B.; et al. Prevalence of diabetes recorded in mainland China using 2018 diagnostic criteria from the American Diabetes Association: National cross sectional study. BMJ 2020, 369, m997. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of nonalcoholic steatohepatitis: An overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Li, H.; Ma, L.; Zhou, J.; Guo, X.; Woo, S.-L.; Pei, Y.; Knight, L.R.; Deveau, M.; Chen, Y.; et al. Expression of STING is increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology 2018, 155, 1971–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Investig. 2019, 129, 546–555. [Google Scholar] [CrossRef]
- Frietze, K.K.; Brown, A.M.; Das, D.; Franks, R.G.; Cunningham, J.L.; Hayward, M.; Nickels, J.T., Jr. Lipotoxicity reduces DDX58/Rig-1 expression and activity leading to impaired autophagy and cell death. Autophagy 2022, 18, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Dolganiuc, A.; Mandrekar, P. Pattern recognition receptors: A contemporary view on liver diseases. Hepatology 2006, 44, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Sato, Y.; Itatsu, K.; Isse, K.; Ikeda, H.; Yasoshima, M.; Zen, Y.; Matsui, A.; Nakanuma, Y. Innate immune response to double-stranded RNA in biliary epithelial cells is associated with the pathogenesis of biliary atresia. Hepatology 2007, 46, 1146–1154. [Google Scholar] [CrossRef]
- Harada, K.; Nakanuma, Y. Cholangiopathy with respect to biliary innate immunity. Int. J. Hepatol. 2012, 2012, 793569. [Google Scholar] [CrossRef] [Green Version]
- Shimada, T.; Imaizumi, T.; Shirai, K.; Tatsuta, T.; Kimura, T.; Hayakari, R.; Yoshida, H.; Matsumiya, T.; Kijima, H.; Mizukami, H.; et al. CCL5 is induced by TLR 3 signaling in HuCCT1 human biliary epithelial cells: Possible involvement in the pathogenesis of biliary atresia. Biomed. Res. 2017, 38, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Chiba, M.; Sasaki, M.; Kitamura, S.; Ikeda, H.; Sato, Y.; Nakanuma, Y. Participation of bile ductular cells in the pathological progression of non-alcoholic fatty liver disease. J. Clin. Pathol. 2011, 64, 564–570. [Google Scholar] [CrossRef]
- Mohs, A.; Otto, T.; Schneider, K.M.; Peltzer, M.; Boekschoten, M.; Holland, C.H.; Hudert, C.A.; Kalveram, L.; Wiegand, S.; Saez-Rodriguez, J.; et al. Hepatocyte-specific NRF2 activation controls fibrogenesis and carcinogenesis in steatohepatitis. J. Hepatol. 2021, 74, 638–648. [Google Scholar] [CrossRef]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, M.S.; Zaninelli, T.H.; Staurengo-Ferrari, L.; Manchope, M.F.; Badaro-Garcia, S.; de Freitas, A.; Casagrande, R.; Verri, W.A. Nrf2 in Immune Responses During Inflammation. In Nrf2 and Its Modulation in Inflammation. Progress in Inflammation Research; Deng, H., Ed.; Springer: Cham, Switzerland, 2020; pp. 23–49. [Google Scholar]
- He, F.; Antonucci, L.; Yamachika, S.; Zhang, Z.; Taniguchi, K.; Umemura, A.; Hatzivassiliou, G.; Roose-Girma, M.; Reina-Campos, M.; Duran, A.; et al. NRF2 activates growth factor genes and downstream AKT signaling to induce mouse and human hepatomegaly. J. Hepatol. 2020, 72, 1182–1195. [Google Scholar] [CrossRef] [Green Version]
- Dash, S.; Aydin, Y.; Moroz, K. Chaperone-mediated autophagy in the liver: Good or bad? Cells 2019, 8, 1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 system in cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [Green Version]
- Engin, A.B.; Engin, A. Obesity and Lipotoxicity; Springer: Cham, Switzerland, 2017; pp. 59–61. [Google Scholar]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-K.J.; Wu, K.C.; Liu, J.; Klaassen, C.D. Nrf2 deficiency improves glucose tolerance in mice fed a high-fat diet. Toxicol. Appl. Pharmacol. 2012, 264, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.A.; Sooro, M.A.; Zhang, P. Autophagic regulation of p62 is critical for cancer therapy. Int. J. Mol. Sci. 2018, 19, 1405. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Barbaro, B.; Romito, I.; Alisi, A. Commentary: The histone demethylase Phf2 acts as a molecular checkpoint to prevent NAFLD progression during obesity. Front. Genet. 2018, 9, 443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brender, C.; Nielsen, M.; Kaltoft, K.; Mikkelsen, G.; Zhang, Q.; Wasik, M.; Billestrup, N.; Odum, N. STAT3-mediated constitutive expression of SOCS-3 in cutaneous T-cell lymphoma. Blood J. Am. Soc. Hematol. 2001, 97, 1056–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim, C.-S.; Jo, K.; Lee, I.S.; Kim, J.-H.; Kim, J.S. POCU1b, the n-butanol soluble fraction of polygoni cuspidati rhizoma et radix, attenuates obesity, non-alcoholic fatty liver, and insulin resistance Via inhibitions of pancreatic lipase, cAMP-dependent PDE activity, AMPK activation, and SOCS-3 suppression. Nutrients 2020, 12, 3612. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.-H.; Chen, X.-Y.; Hu, Q.-H.; Wang, M.-X.; Jin, R.; Zhang, Q.-Y.; Wang, W.; Wang, R.; Kang, L.-L.; et al. Reactive oxygen species-induced TXNIP drives fructose-mediated hepatic inflammation and lipid accumulation through NLRP3 inflammasome activation. Antioxid. Redox Signal. 2015, 22, 848–870. [Google Scholar] [CrossRef] [Green Version]
- Fu, K.; Wang, C.; Ma, C.; Zhou, H.; Li, Y. The potential application of Chinese medicine in liver diseases: A new opportunity. Front. Pharmacol. 2021, 12, 771459. [Google Scholar] [CrossRef] [PubMed]
- Ueki, K.; Kondo, T.; Kahn, C.R. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol. Cell. Biol. 2004, 24, 5434–5446. [Google Scholar] [CrossRef] [Green Version]
- Senn, J.J.; Klover, P.J.; Nowak, I.A.; Zimmers, T.A.; Koniaris, L.G.; Furlanetto, R.W.; Mooney, R.A. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J. Biol. Chem. 2003, 278, 13740–13746. [Google Scholar] [CrossRef] [Green Version]
- Bi, J.; Sun, K.; Wu, H.; Chen, X.; Tang, H.; Mao, J. PPARγ alleviated hepatocyte steatosis through reducing SOCS3 by inhibiting JAK2/STAT3 pathway. Biochem. Biophys. Res. Commun. 2018, 498, 1037–1044. [Google Scholar] [CrossRef]
- Handa, P.; Maliken, B.D.; Nelson, J.E.; Hennessey, K.A.; Vemulakonda, L.A.; Morgan-Stevenson, V.; Dhillon, B.K.; Gupta, R.; Yeh, M.M.; Kowdley, K.V. Differences in hepatic expression of iron, inflammation and stress-related genes in patients with nonalcoholic steatohepatitis. Ann. Hepatol. 2017, 16, 77–85. [Google Scholar] [CrossRef]
- Ogata, H.; Kobayashi, T.; Chinen, T.; Takaki, H.; Sanada, T.; Minoda, Y.; Koga, K.; Takaesu, G.; Maehara, Y.; Iida, M.; et al. Deletion of the SOCS3 gene in liver parenchymal cells promotes hepatitis--induced hepatocarcinogenesis. Gastroenterology 2006, 131, 179–193. [Google Scholar] [CrossRef]
- Sharma, D.; Wang, J.; Fu, P.P.; Sharma, S.; Nagalingam, A.; Mells, J.; Handy, J.; Page, A.J.; Cohen, C.; Anania, F.A.; et al. Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology 2010, 52, 1713–1722. [Google Scholar] [CrossRef] [Green Version]
- Bechmann, L.P.; Kocabayoglu, P.; Sowa, J.-P.; Sydor, S.; Best, J.; Schlattjan, M.; Beilfuss, A.; Schmitt, J.; Hannivoort, R.A.; Kilicarslan, A.; et al. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology 2013, 57, 1394–1406. [Google Scholar] [CrossRef]
- Guo, R.; Nair, S.; Zhang, Y.; Ren, J. Adiponectin deficiency rescues high-fat diet-induced hepatic injury, apoptosis and autophagy loss despite persistent steatosis. Int. J. Obes. 2017, 41, 1403–1412. [Google Scholar] [CrossRef]
- Schöndorf, T.; Maiworm, A.; Emmison, N.; Forst, T.; Pfützner, A. Biological background and role of adiponectin as marker for insulin resistance and cardiovascular risk. Clin. Lab. 2005, 51, 489–494. [Google Scholar]
- Gentile, C.L.; Frye, M.A.; Pagliassotti, M.J. Fatty acids and the endoplasmic reticulum in nonalcoholic fatty liver disease. Biofactors 2011, 37, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Jiao, P.; Ma, J.; Feng, B.; Zhang, H.; Alan-Diehl, J.; Eugene-Chin, Y.; Yan, W.; Xu, H. FFA-induced adipocyte inflammation and insulin resistance: Involvement of ER stress and IKKβ pathways. Obesity 2011, 19, 483–491. [Google Scholar] [CrossRef]
- Nam, T.; Han, J.H.; Devkota, S.; Lee, H.-W. Emerging paradigm of crosstalk between autophagy and the ubiquitin-proteasome system. Mol. Cells 2017, 40, 897–905. [Google Scholar] [PubMed]
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010, 584, 1393–1398. [Google Scholar] [CrossRef] [Green Version]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, U.A.; Kurinna, S.; Schwitter, D.; Marti, A.; Schäfer, M.; Hellerbrand, C.; Speicher, T.; Werner, S. Activated Nrf2 impairs liver regeneration in mice by activation of genes involved in cell-cycle control and apoptosis. Hepatology 2014, 60, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Olagnier, D.; Peri, S.; Steel, C.; van Montfoort, N.; Chiang, C.; Beljanski, V.; Slifker, M.; He, Z.; Nichols, C.N.; Lin, R.; et al. Cellular oxidative stress response controls the antiviral and apoptotic programs in dengue virus-infected dendritic cells. PLoS Pathog. 2014, 10, e1004566. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.J.; Giaccia, A. Dual roles of NRF2 in tumor prevention and progression: Possible implications in cancer treatment. Free Radic. Biol. Med. 2015, 79, 292–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Y.; Lin, W.; Wang, N.; Wei, X.; Huang, Q.; Liao, Y. Bazedoxifene-induced ROS promote mitochondrial dysfunction and enhance osimertinib sensitivity by inhibiting the p-STAT3/SOCS3 and KEAP1/NRF2 pathways in non-small cell lung cancer. Free Radic. Biol. Med. 2023, 196, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Jia, R.-M.; Ji, L.-L.; Qin, X.-M.; Hu, L.; Hu, F.; Han, Y.; Pan, Y.-B.; Jiang, C.-Y.; Liu, W.-T. AMPK-autophagy-mediated inhibition of microRNA-30a-5p alleviates morphine tolerance via SOCS3-dependent neuroinflammation suppression. J. Neuroinflamm. 2022, 19, 25. [Google Scholar] [CrossRef] [PubMed]
- Nakano, D.; Akiba, J.; Tsutsumi, T.; Kawaguchi, M.; Yoshida, T.; Koga, H.; Kawaguchi, T. Hepatic expression of sodium-glucose cotransporter 2 (SGLT2) in patients with chronic liver disease. Med. Mol. Morphol. 2022, 55, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Torisu, T.; Sato, N.; Yoshiga, D.; Kobayashi, T.; Yoshioka, T.; Mori, H.; Iida, M.; Yoshimura, A. The dual function of hepatic SOCS3 in insulin resistance in vivo. Genes Cells 2007, 12, 143–154. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | NAFL (n = 22) | NASH (n = 33) | n Valid Cases | p Value * |
|---|---|---|---|---|
| Age (years) | 38 (24–67) | 45 (20–67) | 22/33 | 0.110 |
| Gender (male/female) | 4/18 | 14/19 | 22/33 | 0.082 |
| BMI (kg/m2) | 49.9 (29.4–66.9) | 53 (27.4–78.19) | 21/31 | 0.208 |
| Adiponectin (µg/mL) | 3.35 (1.3–8.28) | 2.87 (0.83–11.9) | 20/24 | 0.759 |
| CK18 M30 (IU/L) | 174.7 (61.8–807.7) | 366.6 (80.1–1573.9) | 20/24 | 0.002 |
| CK18 M65 (IU/L) | 331.9 (87.9–960.1) | 628.6 (255.8–5273.1) | 19/24 | <0.001 |
| Fasting Glucose (mg/dL) | 95.50 (73–150) | 120 (72–385) | 22/29 | 0.001 |
| Total Cholesterol (mg/dL) | 198 (120–261) | 177.5 (116–247) | 15/18 | 0.320 |
| Triglyceride (mg/dL) | 149 (34–218) | 207 (55–421) | 13/12 | 0.041 |
| ALT (U/L) | 20 (13–65) | 39 (14–120) | 22/31 | 0.001 |
| AST (U/L) | 23 (16–49) | 32.5 (23–90) | 14/20 | <0.001 |
| GGT (U/L) | 20 (2–93) | 43 (15–1213) | 22/31 | <0.001 |
| Fibrosis Grade | ||||
| (0 + 1) | 4 + 7 | 6 + 7 | ||
| (2 + 3) | 10 + 1 | 18 + 2 | 22/33 | 0.58 |
| Steatosis Grade | ||||
| (0 + 1) | 0 + 19 | 0 + 7 | ||
| (2 + 3) | 3 + 0 | 14 + 12 | 22/33 | <0.001 |
| Ballooning Grade | ||||
| (0 + 1) | 17 + 3 | 0 + 16 | ||
| 2 | 2 | 17 | 22/33 | 0.001 |
| Lob. Inflam. Grade | ||||
| (0 + 1) | 16 + 4 | 0 + 11 | ||
| (2 + 3) | 2 + 0 | 20 + 2 | 22/33 | <0.001 |
| pNRF2 | SOCS3 | RIG1 | |||||
|---|---|---|---|---|---|---|---|
| Parameters | n Valid Cases | Median Value b | p Value * | MedianValue b | p Value * | MedianValue b | p Value * |
| Fasting Glucose (mg/dL) | |||||||
| Normal (70–99) | 19 | 0.12 (0–7.25) | 0.29 (0.12–0.45) | 14.31 (2.33–25.25) | |||
| Pre-diabetes and Diabetes a | 32 | 0.69 (0.01–16.93) | 0.029 | 0.33 (0.18–0.45) | 0.090 | 15.98 (3.38–26.62) | 0.205 |
| Total Cholesterol (mg/dL) | |||||||
| Normal (<200) | 20 | 0.27 (0.01–4.06) | 0.3 (0.18–0.45) | 14.45 (3.38–26.28) | |||
| Elevated (>200) | 13 | 0.15 (0–7.25) | 0.957 | 0.31 (0.2–0.45) | 0.813 | 12.14 (3.84–26.62) | 0.548 |
| Triglyceride (mg/dL) | |||||||
| Normal (<200) | 18 | 0.06 (0–1.8) | 0.3 (0.18–0.39) | 12.82 (3.84–23.33) | |||
| Elevated (>200) | 7 | 0.2 (0.01–1.73) | 0.297 | 0.27 (0.2–0.39) | 0.701 | 14.6 (3.38–25.56) | 0.495 |
| ALT (U/L) | |||||||
| Normal (<35/<50) | 35 | 0.29 (0–7.25) | 0.31 (0.12–0.45) | 14.21 (2.33–26.62) | |||
| Elevated (>35/>50) | 18 | 0.67 (0.02–16.93) | 0.195 | 0.34 (0.2–0.57) | 0.276 | 16.29 (5.02–43.11) | 0.244 |
| AST (U/L) | |||||||
| Normal (<35/<50) | 24 | 0.1 (0–3.45) | 0.3 (0.18–0.4) | 14.16 (3.38–25.03) | |||
| Elevated (>35/>50) | 10 | 0.57 (0.1–16.93) | 0.061 | 0.34(0.2–0.57) | 0.223 | 14.53 (5.02–43.11) | 0.539 |
| GGT (U/L) | |||||||
| Normal (<35/<55) | 36 | 0.26 (0–16.93) | 0.32 (0.12–0.57) | 14.26 (2.33–43.11) | |||
| Elevated (>35/>55) | 17 | 0.93 (0.03–5.33) | 0.054 | 0.33 (0.2–0.42) | 0.607 | 16.02 (5.02–26.62) | 0.261 |
| Age Grade | |||||||
| Young (20–40) | 28 | 0.27 (0–14.48) | 0.31 (0.2–0.57) | 14.32 (3.38–43.11) | |||
| Old (41–67) | 27 | 0.74 (0–16.93) | 0.055 | 0.33 (0.12–0.58) | 0.245 | 16.21 (2.33- 54.32) | 0.225 |
| Fibrosis Grade | |||||||
| 0 | 10 | 1.4 (0.18–3.45) | 0.34 (0.28–0.39) | 16.69 (9.59–26.62) | |||
| 1 | 14 | 0.26 (0.04–7.25) | 0.089 | 0.3 (0.12–0.45) | 0.320 | 15.21 (2.33–25.25) | 0.380 |
| 2 | 28 | 0.34 (0–16.93) | 0.091 | 0.32 (0.2–0.58) | 0.619 | 14.94 (3.38–54.32) | 0.691 |
| 3 | 3 | 0.68 (0.21–2.45) | 0.735 | 0.33 (0.33–0.37) | 1.000 | 12.99 (12.3–17.32) | 0.398 |
| Steatosis Grade | |||||||
| 0 + 1 | 26 | 0.11 (0–7.25) | 0.29 (0.12–0.45) | 14.28 (2.33–25.25) | |||
| 2 + 3 | 29 | 0.77 (0–16.93) | 0.011 | 0.35 (0.2–0.58) | 0.008 | 17.03 (3.38–54.32) | 0.032 |
| Ballooning Grade | |||||||
| 0 | 17 | 0.05 (0–0.56) | 0.28 (0.18–0.37) | 12.14 (3.84–20.57) | |||
| 1 | 19 | 0.93 (0.1–16.93) | <0.001 | 0.34 (0.12–0.45) | 0.002 | 15.93 (2.33–25.56) | 0.068 |
| 2 | 19 | 1.42 (0–14.48) | <0.001 | 0.38 (0.2–0.58) | <0.001 | 17.03 (3.38–54.32) | 0.010 |
| Lobular Inflammation Grade | |||||||
| 0 | 16 | 0.09 (0–3.45) | 0.28 (0.12–0.39) | 12.91 (2.33–26.62) | |||
| 1 | 15 | 0.68 (0.05–7.25) | 0.022 | 0.35 (0.18–0.45) | 0.007 | 16.12 (5.02–26.28) | 0.097 |
| 2 | 22 | 0.66 (0–16.93) | 0.051 | 0.36 (0.2–0.58) | 0.007 | 16.42 (3.38–54.32) | 0.174 |
| 3 | 2 | 1.67 (0.1–3.26) | 0.325 | 0.36 (0.33–0.39) | 0.035 | 15.45 (15.1–15.82) | 0.399 |
| Exp (B) | 95% CI | p Value | ||
|---|---|---|---|---|
| Lower | Upper | |||
| A | ||||
| pNRF2 | 2.456 | 1.017 | 5.93 | 0.046 |
| Age (years) | 1.006 | 0.926 | 1.092 | 0.893 |
| BMI (kg/m2) | 1.102 | 0.947 | 1.283 | 0.208 |
| Fibrosis Grade | ||||
| 1 vs. 0 | 1.747 | 0.072 | 42.549 | 0.732 |
| 2 vs. 0 | 1.396 | 0.112 | 17.384 | 0.795 |
| 3 vs. 0 | 0.989 | 0.026 | 38.383 | 0.995 |
| Steatosis Grade | ||||
| (2 + 3) vs. (0 + 1) | 29.267 | 2.237 | 382.895 | 0.01 |
| Ballooning Grade | ||||
| 2 vs. (0 + 1) | 0.628 | 0.041 | 9.674 | 0.739 |
| Lob. Inflam. Grade | ||||
| (2 + 3) vs. (0 + 1) | 19.197 | 1.738 | 211.979 | 0.016 |
| B | ||||
| RIG1 | 1.21 | 1.005 | 1.457 | 0.045 |
| Age (years) | 1.085 | 0.997 | 1.18 | 0.058 |
| BMI (kg/m2) | 1.018 | 0.898 | 1.153 | 0.782 |
| Fibrosis Grade | ||||
| 1 vs. 0 | 2.994 | 0.148 | 60.656 | 0.475 |
| 2 vs. 0 | 1.885 | 0.131 | 27.206 | 0.642 |
| 3 vs. 0 | 0.843 | 0.022 | 32.364 | 0.927 |
| Steatosis Grade | ||||
| (2 + 3) vs. (0 + 1) | 24.689 | 1.788 | 340.962 | 0.017 |
| Ballooning Grade | ||||
| 2 vs. (0 + 1) | 0.384 | 0.021 | 7.164 | 0.521 |
| Lob. Inflam. Grade | ||||
| (2 + 3) vs. (0 + 1) | 35.992 | 2.454 | 527.784 | 0.009 |
| C | ||||
| RIG1 | 1.080 | 0.938 | 1.243 | 0.285 |
| Fasting glucose (mg/dL) | 1.052 | 1.008 | 1.097 | 0.020 |
| Total cholesterol (mg/dL) | 0.995 | 0.972 | 1.018 | 0.682 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwertheim, S.; Alhardan, M.; Manka, P.P.; Sowa, J.-P.; Canbay, A.; Schmidt, H.H.-J.; Baba, H.A.; Kälsch, J. Higher pNRF2, SOCS3, IRF3, and RIG1 Tissue Protein Expression in NASH Patients versus NAFL Patients: pNRF2 Expression Is Concomitantly Associated with Elevated Fasting Glucose Levels. J. Pers. Med. 2023, 13, 1152. https://doi.org/10.3390/jpm13071152
Schwertheim S, Alhardan M, Manka PP, Sowa J-P, Canbay A, Schmidt HH-J, Baba HA, Kälsch J. Higher pNRF2, SOCS3, IRF3, and RIG1 Tissue Protein Expression in NASH Patients versus NAFL Patients: pNRF2 Expression Is Concomitantly Associated with Elevated Fasting Glucose Levels. Journal of Personalized Medicine. 2023; 13(7):1152. https://doi.org/10.3390/jpm13071152
Chicago/Turabian StyleSchwertheim, Suzan, Malek Alhardan, Paul P. Manka, Jan-Peter Sowa, Ali Canbay, Hartmut H.-J. Schmidt, Hideo A. Baba, and Julia Kälsch. 2023. "Higher pNRF2, SOCS3, IRF3, and RIG1 Tissue Protein Expression in NASH Patients versus NAFL Patients: pNRF2 Expression Is Concomitantly Associated with Elevated Fasting Glucose Levels" Journal of Personalized Medicine 13, no. 7: 1152. https://doi.org/10.3390/jpm13071152