
Integrated Bioinformatics Investigation of Novel Biomarkers of Uterine Leiomyosarcoma Diagnosis and Outcome

Abstract
:1. Introduction
2. Materials and Methods
2.1. Microarray Data Mining and Identification of DEGs
2.2. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analysis
2.3. Protein-Protein Interaction (PPI) Network Construction
2.4. Identification of Hub Genes
2.5. Validation of Hub Genes and Survival Analysis
2.6. TFs and miRNAs Related to Hub Genes
2.7. Drug–Hub Gene Interaction
3. Results
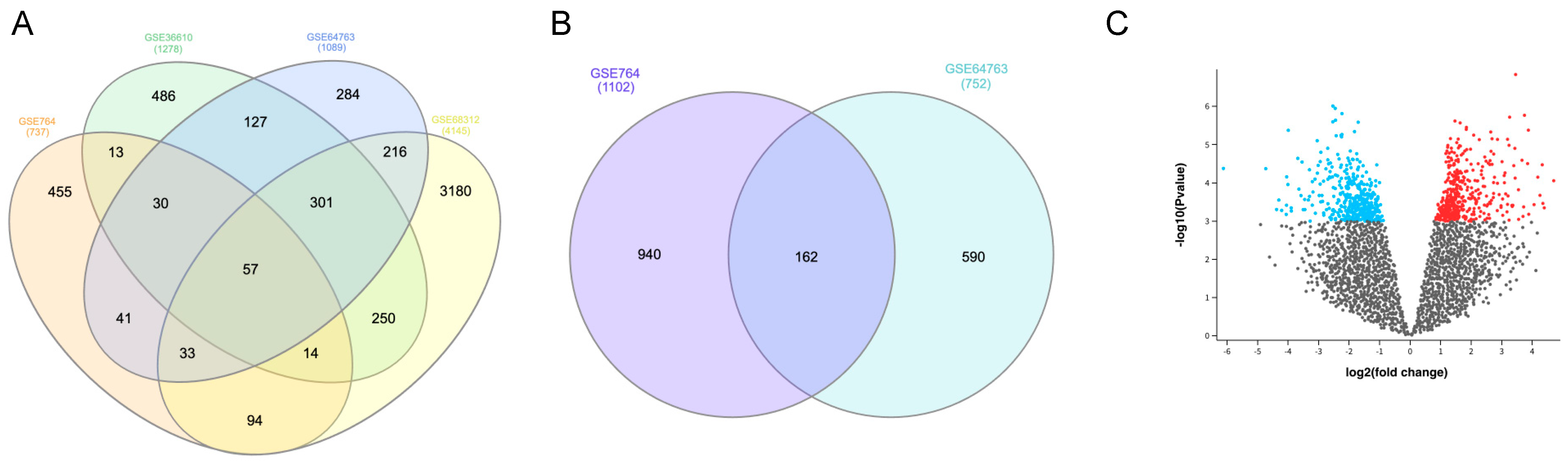
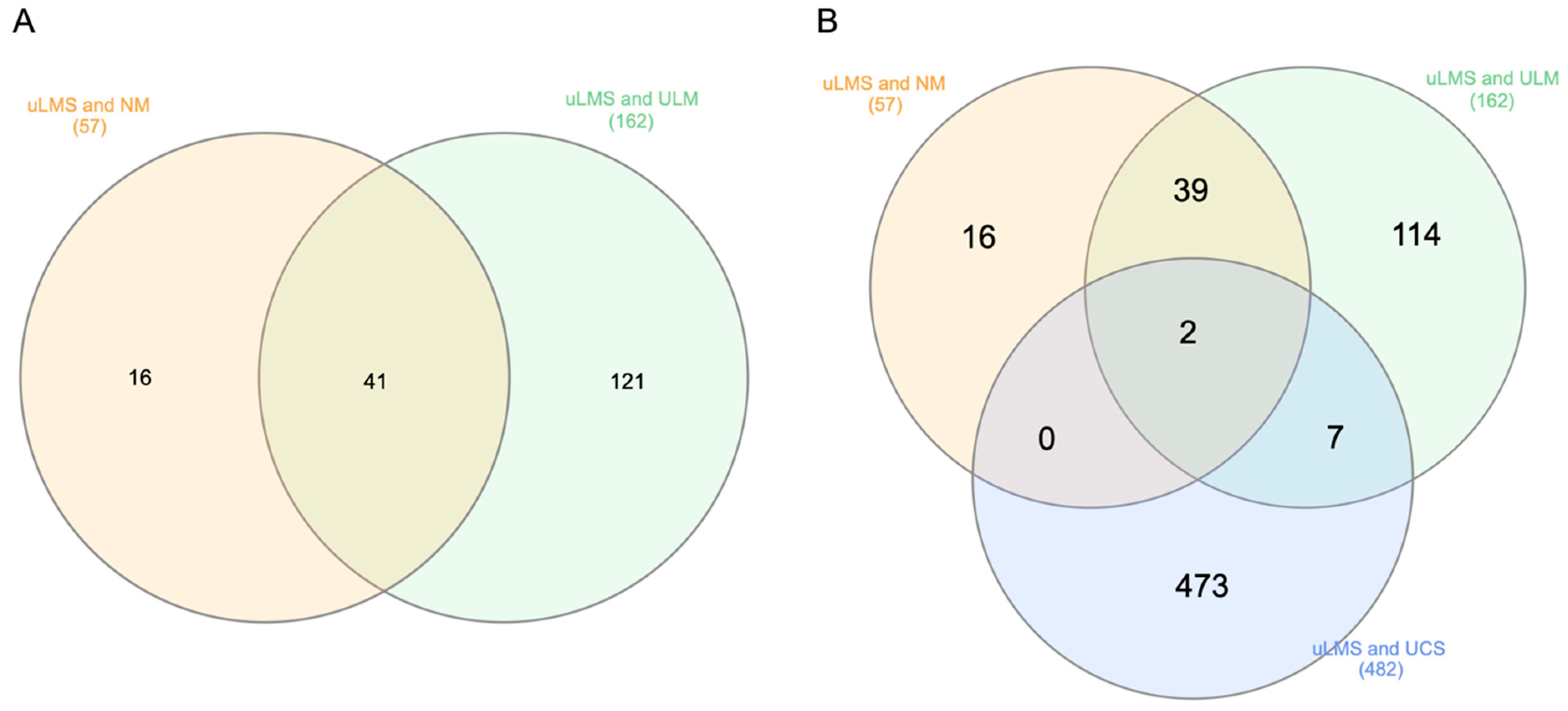
3.1. Identification of DEGs
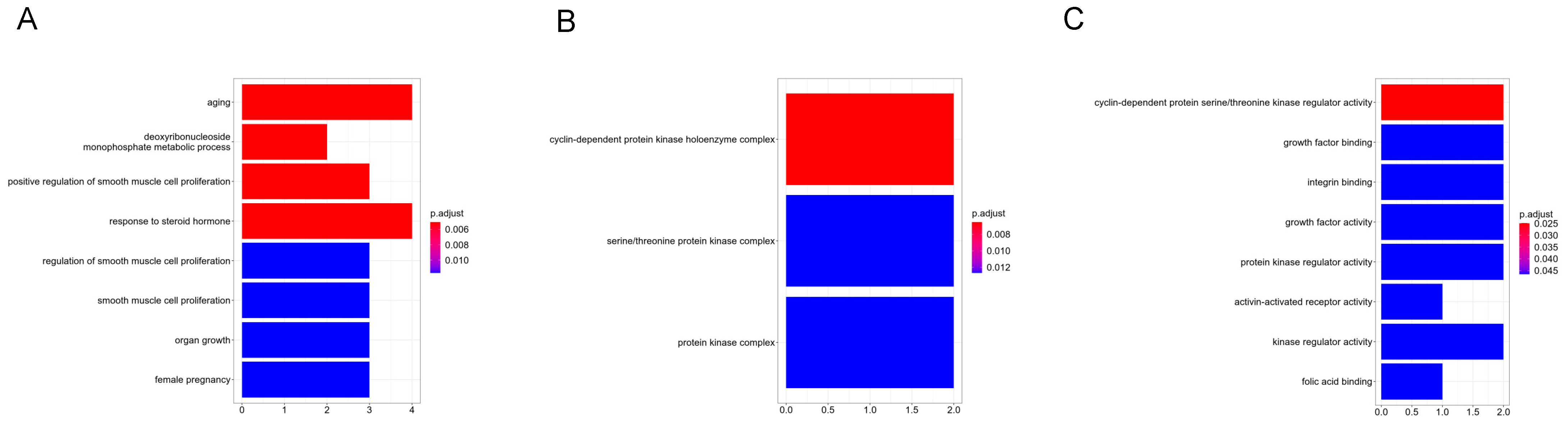
3.2. GO and KEGG Pathway Enrichment Analysis
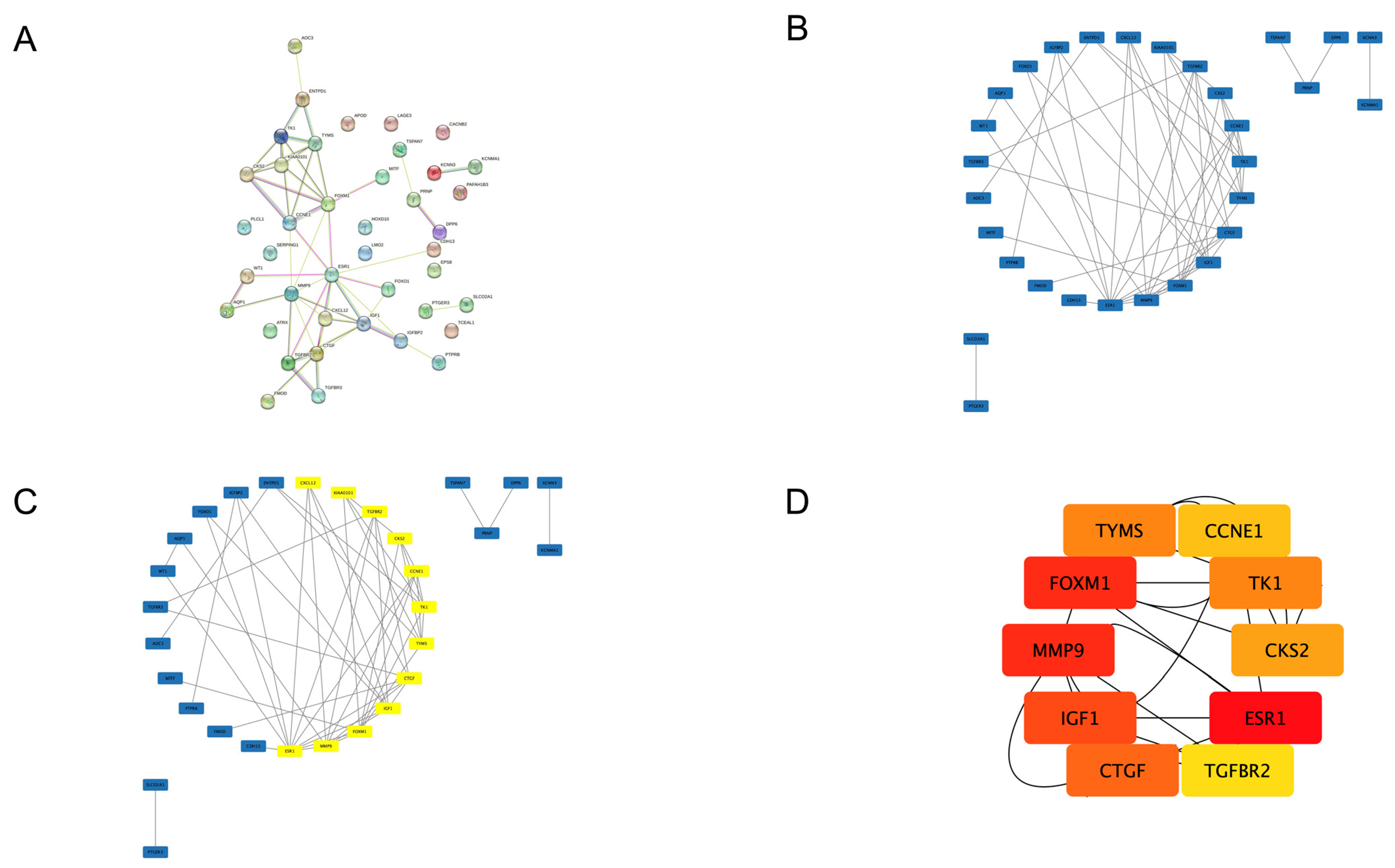
3.3. PPI Network and Hub Gene Selection
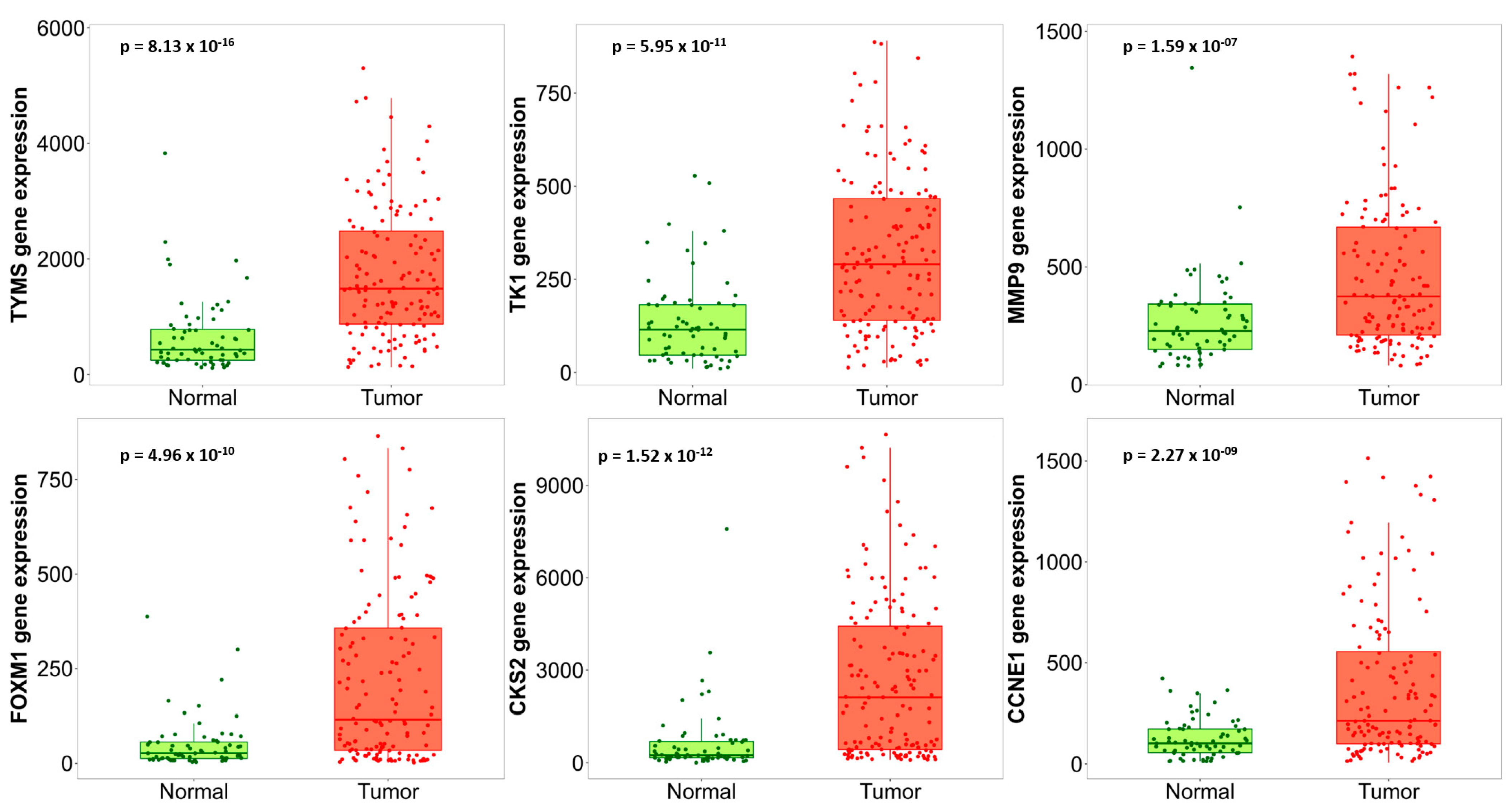
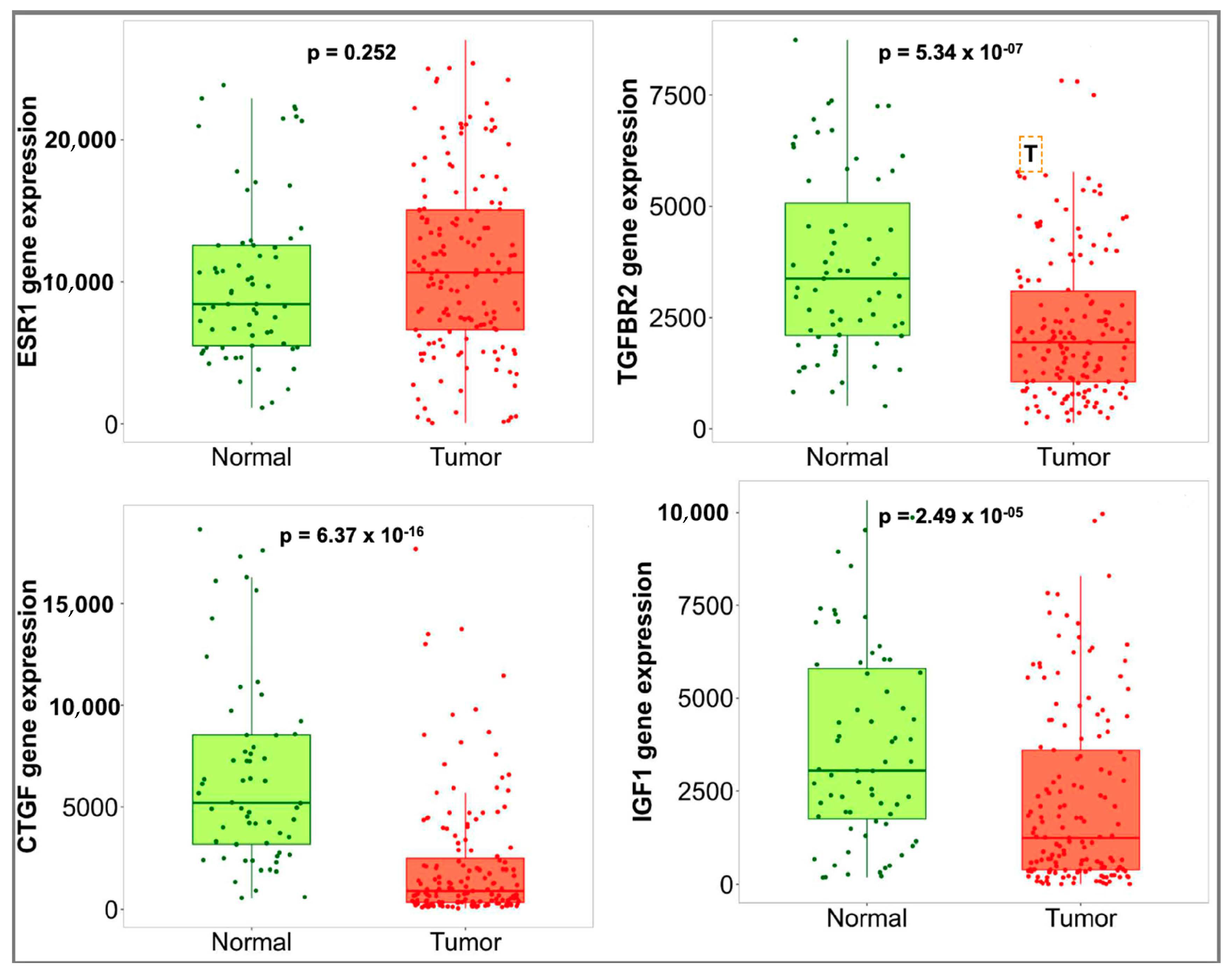
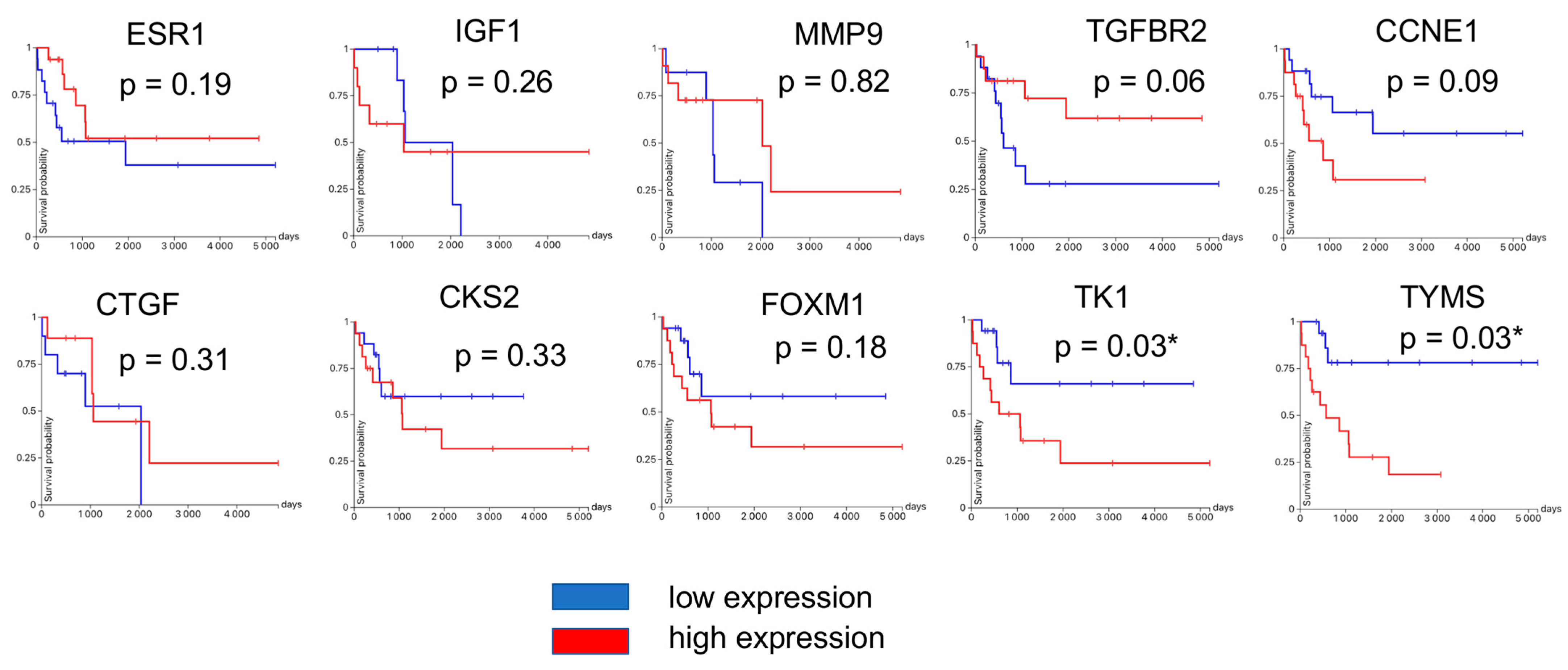
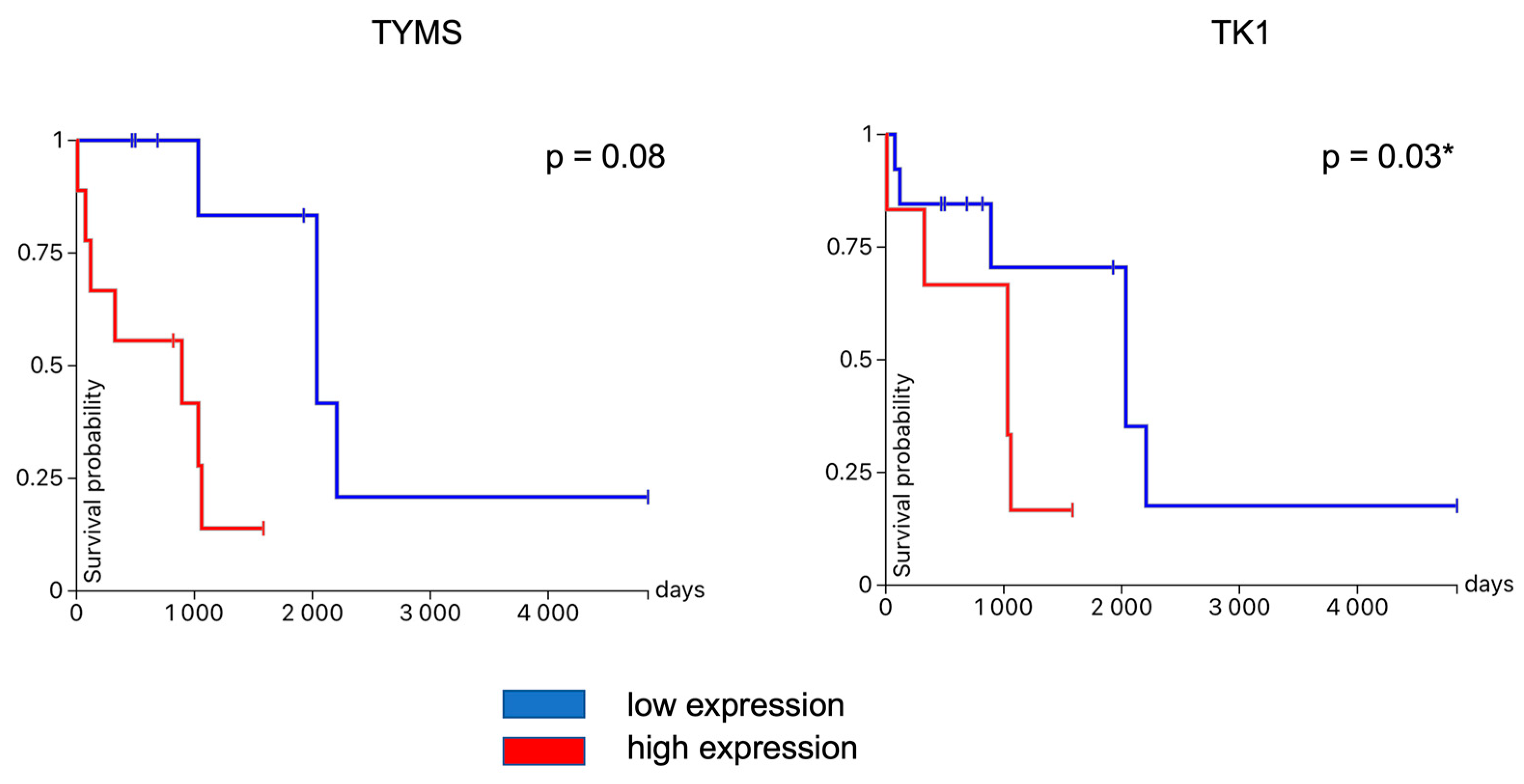
3.4. Hub Gene Validation and Survival Analysis
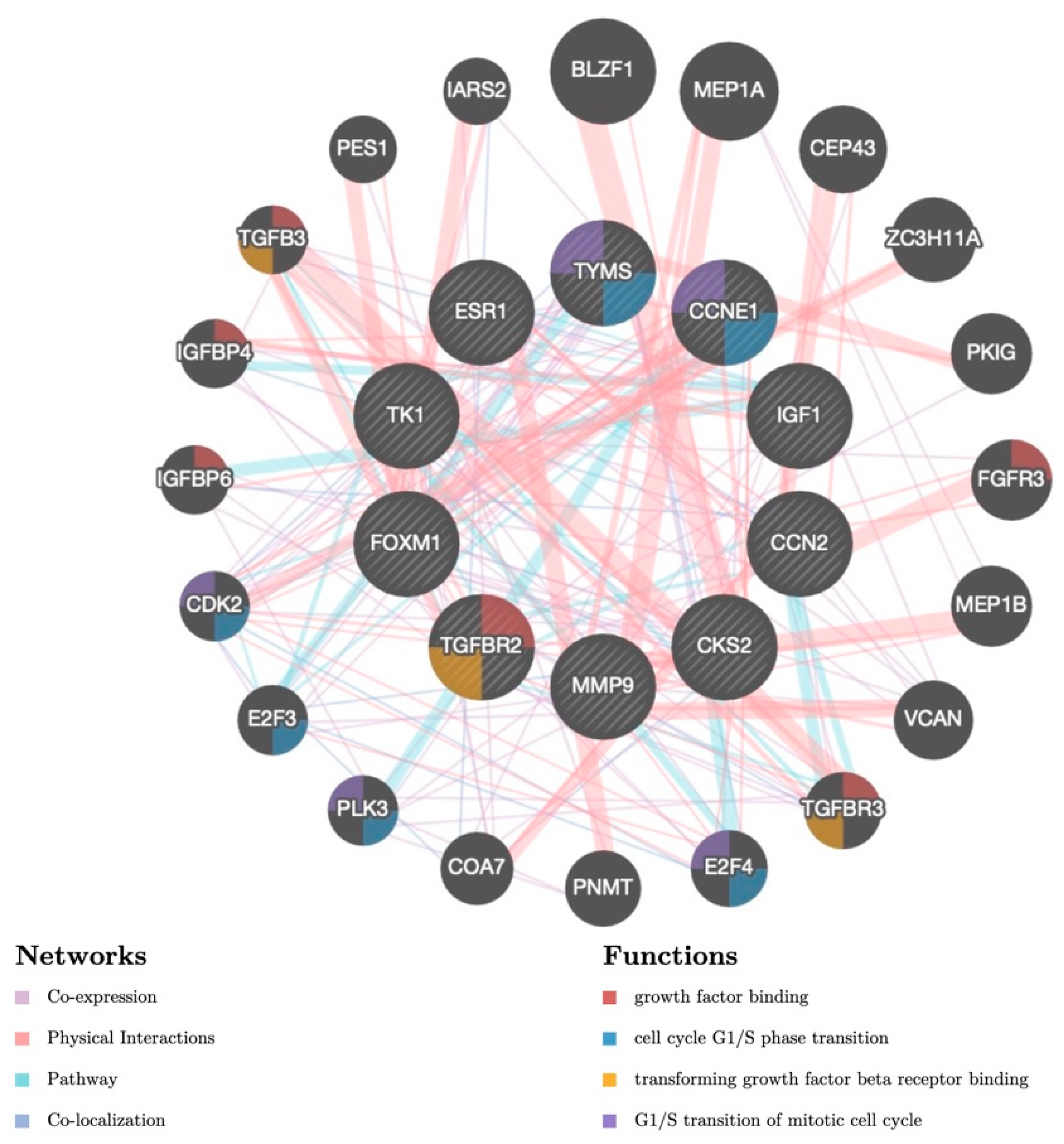
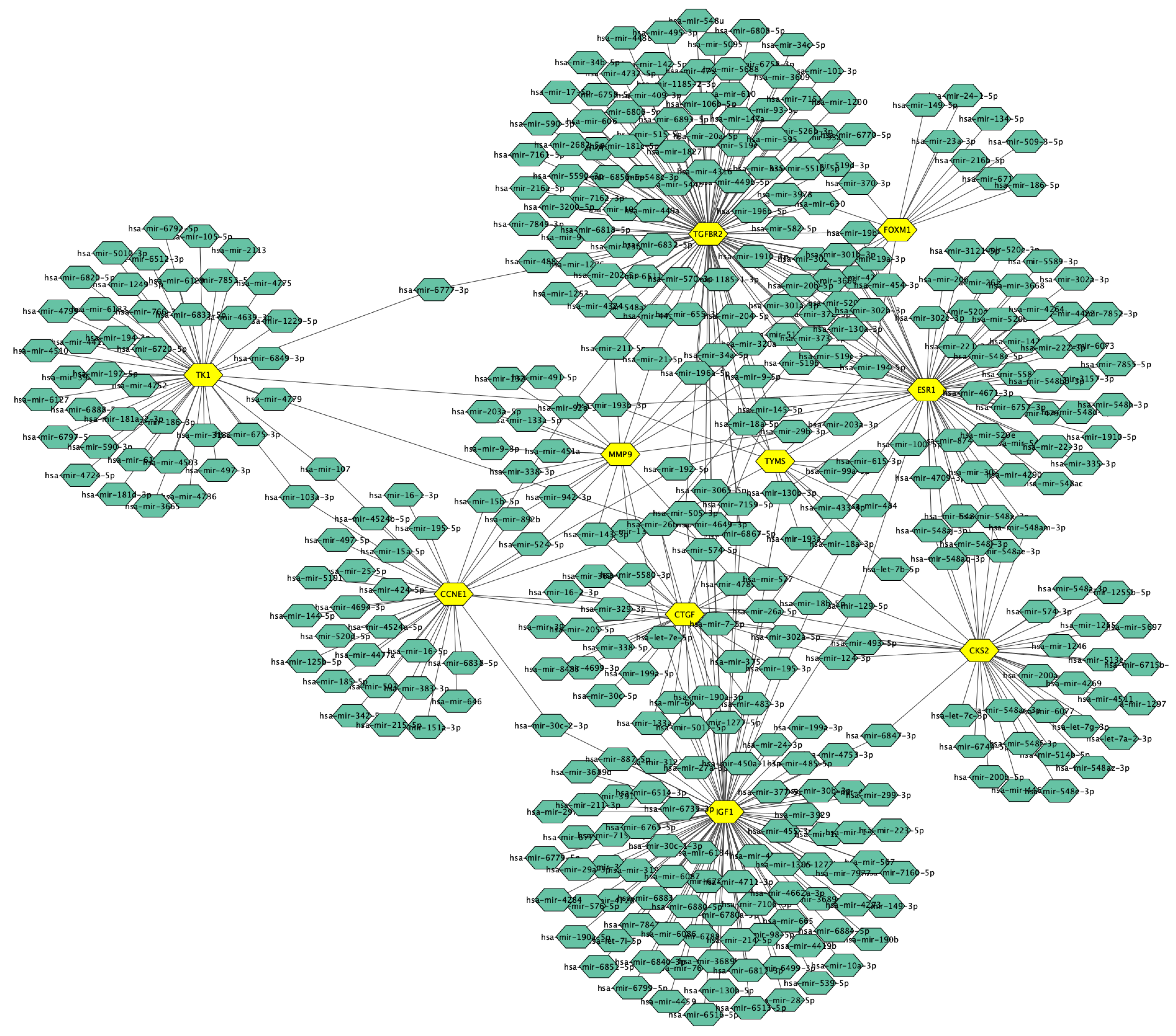
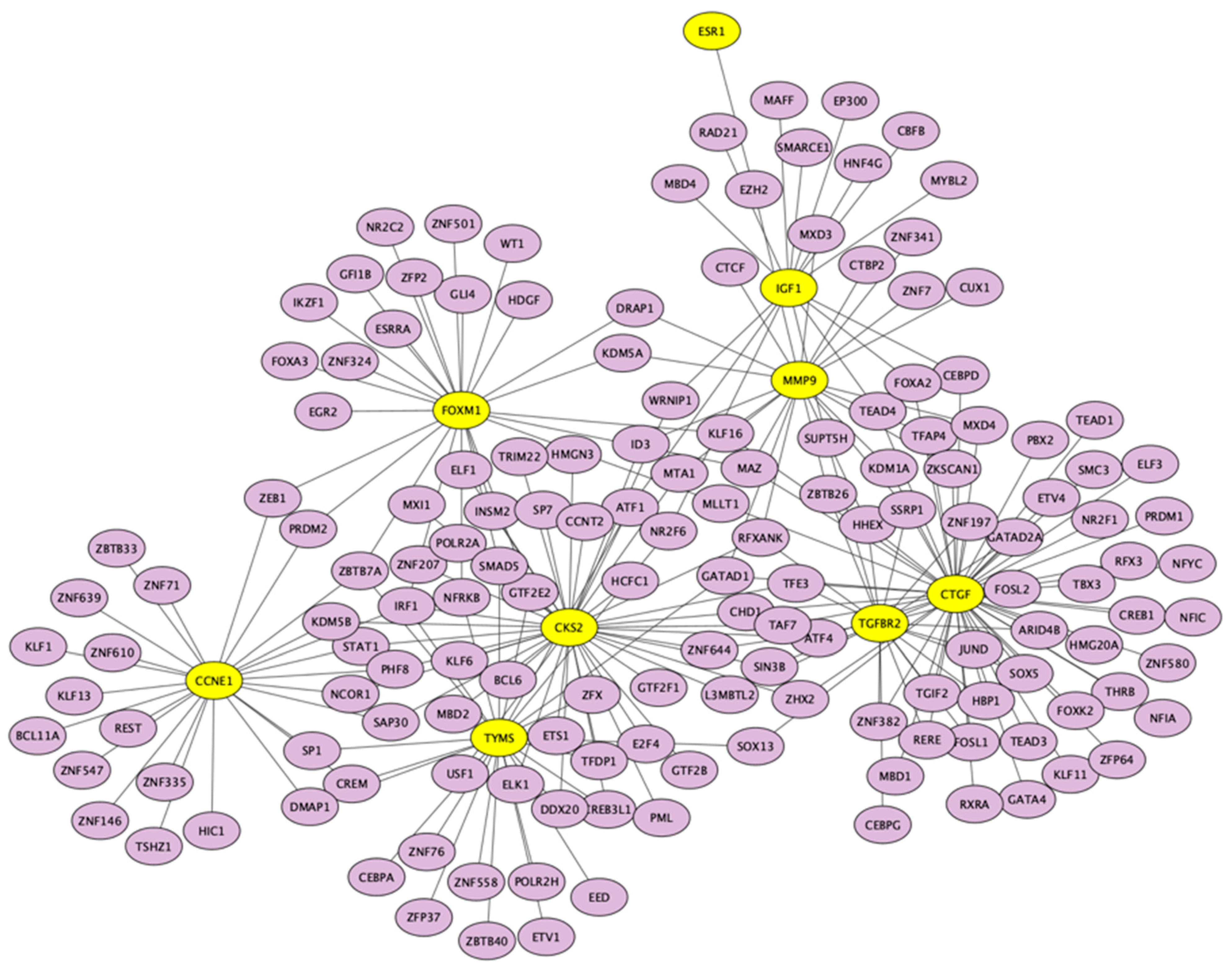
3.5. Candidate TFs and miRNAs Related to Hub Genes
3.6. Drug-Gene Interaction Network
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roberts, M.E.; Aynardi, J.T.; Chu, C.S. Uterine leiomyosarcoma: A review of the literature and update on management options. Gynecol. Oncol. 2018, 151, 562–572. [Google Scholar] [CrossRef]
- Hosh, M.; Antar, S.; Nazzal, A.; Warda, M.; Gibreel, A.; Refky, B. Uterine Sarcoma: Analysis of 13,089 Cases Based on Surveillance, Epidemiology, and End Results Database. Int. J. Gynecol. Cancer 2016, 26, 1098–1104. [Google Scholar] [CrossRef]
- Juhasz-Böss, I.; Gabriel, L.; Bohle, R.M.; Horn, L.C.; Solomayer, E.-F.; Breitbach, G.-P. Uterine Leiomyosarcoma. Oncol. Res. Treat. 2018, 41, 680–686. [Google Scholar] [CrossRef]
- de Almeida, B.C.; dos Anjos, L.G.; Dobroff, A.S.; Baracat, E.C.; Yang, Q.; Al-Hendy, A.; Carvalho, K.C. Epigenetic Features in Uterine Leiomyosarcoma and Endometrial Stromal Sarcomas: An Overview of the Literature. Biomedicines 2022, 10, 2567. [Google Scholar] [CrossRef]
- Sun, S.; Bonaffini, P.A.; Nougaret, S.; Fournier, L.; Dohan, A.; Chong, J.; Smith, J.; Addley, H.; Reinhold, C. How to differentiate uterine leiomyosarcoma from leiomyoma with imaging. Diagn. Interv. Imaging 2019, 100, 619–634. [Google Scholar] [CrossRef]
- Kaganov, H.; Ades, A.; Fraser, D.S. Preoperative Magnetic Resonance Imaging Diagnostic Features of Uterine Leiomyosarcomas: A Systematic Review. Int. J. Technol. Assess. Health Care 2018, 34, 172–179. [Google Scholar] [CrossRef]
- Seagle, B.-L.L.; Sobecki-Rausch, J.; Strohl, A.E.; Shilpi, A.; Grace, A.; Shahabi, S. Prognosis and treatment of uterine leiomyosarcoma: A National Cancer Database study. Gynecol. Oncol. 2017, 145, 61–70. [Google Scholar] [CrossRef]
- Asano, H.; Isoe, T.; Ito, Y.M.; Nishimoto, N.; Watanabe, Y.; Yokoshiki, S.; Watari, H. Status of the Current Treatment Options and Potential Future Targets in Uterine Leiomyosarcoma: A Review. Cancers 2022, 14, 1180. [Google Scholar] [CrossRef]
- Reed, N.S.; Mangioni, C.; Malmström, H.; Scarfone, G.; Poveda, A.; Pecorelli, S.; Tateo, S.; Franchi, M.; Jobsen, J.J.; Coens, C.; et al. Phase III randomised study to evaluate the role of adjuvant pelvic radiotherapy in the treatment of uterine sarcomas stages I and II: An European Organisation for Research and Treatment of Cancer Gynaecological Cancer Group Study (protocol 55874). Eur. J. Cancer 2008, 44, 808–818. [Google Scholar] [CrossRef]
- Chae, S.H.; Shim, S.-H.; Chang, M.; Choi, A.Y.; Kang, G.G.; Lee, S.J.; Kim, S.-N. Effect of adjuvant therapy on the risk of recurrence in early-stage leiomyosarcoma: A meta-analysis. Gynecol. Oncol. 2019, 154, 638–650. [Google Scholar] [CrossRef]
- Athieniti, E.; Spyrou, G.M. A guide to multi-omics data collection and integration for translational medicine. Comput. Struct. Biotechnol. J. 2023, 21, 134–149. [Google Scholar] [CrossRef]
- Abbas-Aghababazadeh, F.; Mo, Q.; Fridley, B.L. Statistical genomics in rare cancer. Semin. Cancer Biol. 2020, 61, 1–10. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. In Statistical Genomics: Methods and Protocols; Mathé, E., Davis, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 93–110. ISBN 978-1-4939-3578-9. [Google Scholar]
- Wang, Z.; Lachmann, A.; Ma’ayan, A. Mining data and metadata from the gene expression omnibus. Biophys. Rev. 2019, 11, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Ganini, C.; Amelio, I.; Bertolo, R.; Bove, P.; Buonomo, O.C.; Candi, E.; Cipriani, C.; Di Daniele, N.; Juhl, H.; Mauriello, A.; et al. Global mapping of cancers: The Cancer Genome Atlas and beyond. Mol. Oncol. 2021, 15, 2823–2840. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A Web-Based Tool for the Analysis of Sets through Venn Diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology Consortium. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2–27. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [Green Version]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [Green Version]
- Bartha, Á.; Győrffy, B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci. 2021, 22, 2622. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Pannier, D.; Cordoba, A.; Ryckewaert, T.; Robin, Y.-M.; Penel, N. Hormonal therapies in uterine sarcomas, aggressive angiomyxoma, and desmoid-type fibromatosis. Crit. Rev. Oncol./Hematol. 2019, 143, 62–66. [Google Scholar] [CrossRef]
- Maccaroni, E.; Lunerti, V.; Agostinelli, V.; Giampieri, R.; Zepponi, L.; Pagliacci, A.; Berardi, R. New Insights into Hormonal Therapies in Uterine Sarcomas. Cancers 2022, 14, 921. [Google Scholar] [CrossRef]
- Ioffe, Y.J.; Li, A.J.; Walsh, C.S.; Karlan, B.Y.; Leuchter, R.; Forscher, C.; Cass, I. Hormone receptor expression in uterine sarcomas: Prognostic and therapeutic roles. Gynecol. Oncol. 2009, 115, 466–471. [Google Scholar] [CrossRef]
- Leitao, M.M.; Soslow, R.A.; Nonaka, D.; Olshen, A.B.; Aghajanian, C.; Sabbatini, P.; Dupont, J.; Hensley, M.; Sonoda, Y.; Barakat, R.R.; et al. Tissue microarray immunohistochemical expression of estrogen, progesterone, and androgen receptors in uterine leiomyomata and leiomyosarcoma. Cancer 2004, 101, 1455–1462. [Google Scholar] [CrossRef]
- Abeshouse, A.; Adebamowo, C.; Adebamowo, S.N.; Akbani, R.; Akeredolu, T.; Ally, A.; Anderson, M.L.; Anur, P.; Appelbaum, E.L.; Armenia, J.; et al. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar] [CrossRef] [Green Version]
- Koivisto-Korander, R.; Butzow, R.; Koivisto, A.-M.; Leminen, A. Immunohistochemical studies on uterine carcinosarcoma, leiomyosarcoma, and endometrial stromal sarcoma: Expression and prognostic importance of ten different markers. Tumor Biol. 2011, 32, 451–459. [Google Scholar] [CrossRef]
- Zang, Y.; Dong, M.; Zhang, K.; Gao, C.; Guo, F.; Wang, Y.; Xue, F. Hormonal therapy in uterine sarcomas. Cancer Med. 2019, 8, 1339–1349. [Google Scholar] [CrossRef] [Green Version]
- George, S.; Feng, Y.; Manola, J.; Nucci, M.R.; Butrynski, J.E.; Morgan, J.A.; Ramaiya, N.; Quek, R.; Penson, R.T.; Wagner, A.J.; et al. Phase 2 trial of aromatase inhibition with letrozole in patients with uterine leiomyosarcomas expressing estrogen and/or progesterone receptors. Cancer 2014, 120, 738–743. [Google Scholar] [CrossRef]
- Huang, H. Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors 2018, 18, 3249. [Google Scholar] [CrossRef] [Green Version]
- Gobin, E.; Bagwell, K.; Wagner, J.; Mysona, D.; Sandirasegarane, S.; Smith, N.; Bai, S.; Sharma, A.; Schleifer, R.; She, J.-X. A pan-cancer perspective of matrix metalloproteases (MMP) gene expression profile and their diagnostic/prognostic potential. BMC Cancer 2019, 19, 581. [Google Scholar] [CrossRef] [Green Version]
- Mehner, C.; Hockla, A.; Miller, E.; Ran, S.; Radisky, D.C.; Radisky, E.S. Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget 2014, 5, 2736–2749. [Google Scholar] [CrossRef] [Green Version]
- Mondal, S.; Adhikari, N.; Banerjee, S.; Amin, S.A.; Jha, T. Matrix metalloproteinase-9 (MMP-9) and its inhibitors in cancer: A minireview. Eur. J. Med. Chem. 2020, 194, 112260. [Google Scholar] [CrossRef]
- Pego, E.R.; Fernández, I.; Núñez, M.J. Molecular basis of the effect of MMP-9 on the prostate bone metastasis: A review. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 272–282. [Google Scholar] [CrossRef]
- Zeng, Y.; Gao, M.; Lin, D.; Du, G.; Cai, Y. Prognostic and Immunological Roles of MMP-9 in Pan-Cancer. BioMed. Res. Int. 2022, 2022, 2592962. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, X.; Zhang, Y.; Qiao, J.; Sasano, H.; McNamara, K.; Zhao, B.; Zhang, D.; Fan, Y.; Liu, L.; et al. Estradiol-Induced MMP-9 Expression via PELP1-Mediated Membrane-Initiated Signaling in ERα-Positive Breast Cancer Cells. Horm. Cancer 2020, 11, 87–96. [Google Scholar] [CrossRef]
- Liao, G.-B.; Li, X.-Z.; Zeng, S.; Liu, C.; Yang, S.-M.; Yang, L.; Hu, C.-J.; Bai, J.-Y. Regulation of the master regulator FOXM1 in cancer. Cell Commun. Signal. 2018, 16, 57. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wu, D.; Yu, Q.; Li, L.; Wu, P. Prognostic value of FOXM1 in solid tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 32298–32308. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Chen, L.; Yie, L.; Wei, L.; Wen, T.; Liu, Y.; Chen, H. Targeting FoxM1 inhibits proliferation, invasion and migration of nasopharyngeal carcinoma through the epithelial-to-mesenchymal transition pathway. Oncol. Rep. 2015, 33, 2402–2410. [Google Scholar] [CrossRef] [Green Version]
- Martinsson-Ahlzén, H.-S.; Liberal, V.; Grünenfelder, B.; Chaves, S.R.; Spruck, C.H.; Reed, S.I. Cyclin-Dependent Kinase-Associated Proteins Cks1 and Cks2 Are Essential during Early Embryogenesis and for Cell Cycle Progression in Somatic Cells. Mol. Cell Biol. 2008, 28, 5698–5709. [Google Scholar] [CrossRef] [Green Version]
- Spruck, C.H.; de Miguel, M.P.; Smith, A.P.L.; Ryan, A.; Stein, P.; Schultz, R.M.; Lincoln, A.J.; Donovan, P.J.; Reed, S.I. Requirement of Cks2 for the First Metaphase/Anaphase Transition of Mammalian Meiosis. Science 2003, 300, 647–650. [Google Scholar] [CrossRef]
- Huang, N.; Wu, Z.; Hong, H.; Wang, X.; Yang, F.; Li, H. Overexpression of CKS2 is associated with a poor prognosis and promotes cell proliferation and invasion in breast cancer. Mol. Med. Rep. 2019, 19, 4761–4769. [Google Scholar] [CrossRef]
- Kang, M.A.; Kim, J.-T.; Kim, J.H.; Kim, S.-Y.; Kim, Y.H.; Yeom, Y.I.; Lee, Y.; Lee, H.G. Upregulation of the cycline kinase subunit CKS2 increases cell proliferation rate in gastric cancer. J. Cancer Res. Clin. Oncol. 2009, 135, 761–769. [Google Scholar] [CrossRef]
- Yu, M.; Zhong, M.; Qiao, Z. Expression and clinical significance of cyclin kinase subunit 2 in colorectal cancer. Oncol. Lett. 2013, 6, 777–780. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Song, Q.; Liu, J.; Lu, L.; Xu, Y.; Zheng, W. Cyclin-Dependent Kinase Regulatory Subunit 2 Indicated Poor Prognosis and Facilitated Aggressive Phenotype of Hepatocellular Carcinoma. Dis. Markers 2019, 2019, 8964015. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Han, Q.; Mei, S.; Li, H.; Yang, F.; Wang, J.; Ge, S.; Jing, X.; Xu, H.; Zhang, T. Cyclin-dependent kinase subunit 2 overexpression promotes tumor progression and predicts poor prognosis in uterine leiomyosarcoma. Oncol. Lett. 2019, 18, 2845–2852. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, P.; López-Contreras, A.J. ATRX, a guardian of chromatin. Trends Genet. 2023, 39, 505–519. [Google Scholar] [CrossRef]
- Juhász, S.; Elbakry, A.; Mathes, A.; Löbrich, M. ATRX Promotes DNA Repair Synthesis and Sister Chromatid Exchange during Homologous Recombination. Mol. Cell 2018, 71, 11–24.e7. [Google Scholar] [CrossRef] [Green Version]
- Lovejoy, C.A.; Takai, K.; Huh, M.S.; Picketts, D.J.; de Lange, T. ATRX affects the repair of telomeric DSBs by promoting cohesion and a DAXX-dependent activity. PLOS Biol. 2020, 18, e3000594. [Google Scholar] [CrossRef] [Green Version]
- Voon, H.P.J.; Hughes, J.R.; Rode, C.; De La Rosa-Velázquez, I.A.; Jenuwein, T.; Feil, R.; Higgs, D.; Gibbons, R.J. ATRX Plays a Key Role in Maintaining Silencing at Interstitial Heterochromatic Loci and Imprinted Genes. Cell Rep. 2015, 11, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Tu, C.; Tang, Z.; Ma, R.; Li, Z. Alternative lengthening of telomeres phenotype and loss of ATRX expression in sarcomas (Review). Oncol. Lett. 2018, 15, 7489–7496. [Google Scholar] [CrossRef] [Green Version]
- De Vitis, M.; Berardinelli, F.; Sgura, A. Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT). Int. J. Mol. Sci. 2018, 19, 606. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Deng, Z.; Zhang, L.; Wu, C.; Jin, Y.; Hwang, I.; Vladimirova, O.; Xu, L.; Yang, L.; Lu, B.; et al. ATRX loss induces telomere dysfunction and necessitates induction of alternative lengthening of telomeres during human cell immortalization. EMBO J. 2019, 38, e96659. [Google Scholar] [CrossRef]
- Choi, J.; Manzano, A.; Dong, W.; Bellone, S.; Bonazzoli, E.; Zammataro, L.; Yao, X.; Deshpande, A.; Zaidi, S.; Guglielmi, A.; et al. Integrated mutational landscape analysis of uterine leiomyosarcomas. Proc. Natl. Acad. Sci. USA 2021, 118, 2025182118. [Google Scholar] [CrossRef]
- Tsuyoshi, H.; Yoshida, Y. Molecular biomarkers for uterine leiomyosarcoma and endometrial stromal sarcoma. Cancer Sci. 2018, 109, 1743–1752. [Google Scholar] [CrossRef]
- Xie, H.; Guo, L.; Wang, Z.; Peng, S.; Ma, Q.; Yang, Z.; Shang, Z.; Niu, Y. Assessing the Potential Prognostic and Immunological Role of TK1 in Prostate Cancer. Front. Genet. 2022, 13, 778850. [Google Scholar] [CrossRef]
- Fanelli, G.N.; Scarpitta, R.; Cinacchi, P.; Fuochi, B.; Szumera-Ciećkiewicz, A.; De Ieso, K.; Ferrari, P.; Fontana, A.; Miccoli, M.; Naccarato, A.G.; et al. Immunohistochemistry for Thymidine Kinase-1 (TK1): A Potential Tool for the Prognostic Stratification of Breast Cancer Patients. J. Clin. Med. 2021, 10, 5416. [Google Scholar] [CrossRef]
- Bitter, E.E.; Townsend, M.H.; Erickson, R.; Allen, C.; O’neill, K.L. Thymidine kinase 1 through the ages: A comprehensive review. Cell Biosci. 2020, 10, 138. [Google Scholar] [CrossRef]
- Xu, Y.; Shi, Q.-L.; Ma, H.; Zhou, H.; Lu, Z.; Yu, B.; Zhou, X.; Eriksson, S.; He, E.; Skog, S. High thymidine kinase 1 (TK1) expression is a predictor of poor survival in patients with pT1 of lung adenocarcinoma. Tumor Biol. 2012, 33, 475–483. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, D.; Li, D.; Li, D.; Wang, H.; Wu, Y. Clinical Value on Combined Detection of Serum CA724, DKK1, and TK1 in Diagnosis of Gastric Cancer. J. Oncol. 2022, 2022, 6941748. [Google Scholar] [CrossRef]
- Malvi, P.; Janostiak, R.; Nagarajan, A.; Cai, G.; Wajapeyee, N. Loss of thymidine kinase 1 inhibits lung cancer growth and metastatic attributes by reducing GDF15 expression. PLOS Genet. 2019, 15, e1008439. [Google Scholar] [CrossRef] [Green Version]
- Nisman, B.; Allweis, T.; Kaduri, L.; Maly, B.; Gronowitz, S.; Hamburger, T.; Peretz, T. Serum thymidine kinase 1 activity in breast cancer. Cancer Biomarkers 2010, 7, 65–72. [Google Scholar] [CrossRef]
- Wei, Y.-T.; Luo, Y.-Z.; Feng, Z.-Q.; Huang, Q.-X.; Mo, A.-S.; Mo, S.-X. TK1 overexpression is associated with the poor outcomes of lung cancer patients: A systematic review and meta-analysis. Biomarkers Med. 2018, 12, 403–413. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, X.; Dong, S.; Shen, J.; Yu, H.; Zhou, J.; Li, J.; Ma, H.; He, E.; Skog, S. Serum TK1 is a more reliable marker than CEA and AFP for cancer screening in a study of 56,286 people. Cancer Biomarkers 2016, 16, 529–536. [Google Scholar] [CrossRef] [Green Version]
- Burdelski, C.; Strauss, C.; Tsourlakis, M.C.; Kluth, M.; Hube-Magg, C.; Melling, N.; Lebok, P.; Minner, S.; Koop, C.; Graefen, M.; et al. Overexpression of thymidylate synthase (TYMS) is associated with aggressive tumor features and early PSA recurrence in prostate cancer. Oncotarget 2015, 6, 8377–8387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, L.; Voeller, D.; Rahman, M.; Lipkowitz, S.; Allegra, C.; Barrett, J.C.; Kaye, F.J.; Zajac-Kaye, M. Thymidylate synthase as an oncogene: A novel role for an essential DNA synthesis enzyme. Cancer Cell 2004, 5, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Jiao, Y.; Li, Y.; Ji, B.; Jia, B.; Liu, B. TYMS presents a novel biomarker for diagnosis and prognosis in patients with pancreatic cancer. Medicine 2019, 98, e18487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yan, L.; Cui, C.; Wang, Z.; Wu, J.; Zhao, M.; Dong, B.; Guan, X.; Tian, X.; Hao, C. Identification of TYMS as a promoting factor of retroperitoneal liposarcoma progression: Bioinformatics analysis and biological evidence. Oncol. Rep. 2020, 44, 565–576. [Google Scholar] [CrossRef]
- Mannherz, W.; Agarwal, S. Thymidine nucleotide metabolism controls human telomere length. Nat. Genet. 2023, 55, 568–580. [Google Scholar] [CrossRef]
- Vizcaíno, C.; Mansilla, S.; Portugal, J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacol. Ther. 2015, 152, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Dauer, P.; Gupta, V.K.; McGinn, O.; Nomura, A.; Sharma, N.S.; Arora, N.; Giri, B.; Dudeja, V.; Saluja, A.K.; Banerjee, S. Inhibition of Sp1 prevents ER homeostasis and causes cell death by lysosomal membrane permeabilization in pancreatic cancer. Sci. Rep. 2017, 7, 1564. [Google Scholar] [CrossRef] [Green Version]
- Beishline, K.; Azizkhan-Clifford, J. Sp1 and the ‘hallmarks of cancer’. FEBS J. 2015, 282, 224–258. [Google Scholar] [CrossRef]
- Ning, R.; Meng, S.; Wang, L.; Jia, Y.; Tang, F.; Sun, H.; Zhang, Z.; Zhang, C.; Fan, X.; Xiao, B.; et al. 6 Circulating miRNAs can be used as Non-invasive Biomarkers for the Detection of Cervical Lesions. J. Cancer 2021, 12, 5106–5113. [Google Scholar] [CrossRef]
- Li, M.; Xiao, Y.; Liu, M.; Ning, Q.; Xiang, Z.; Zheng, X.; Tang, S.; Mo, Z. MiR-26a-5p regulates proliferation, apoptosis, migration and invasion via inhibiting hydroxysteroid dehydrogenase like-2 in cervical cancer cell. BMC Cancer 2022, 22, 876. [Google Scholar] [CrossRef]
- Friedman, C.F.; Hensley, M.L. Options for Adjuvant Therapy for Uterine Leiomyosarcoma. Curr. Treat. Options Oncol. 2018, 19, 7. [Google Scholar] [CrossRef]
- Takehara, K.; Yamashita, N.; Watanabe, R.; Teramoto, N.; Tsuda, H.; Motohashi, T.; Harano, K.; Nakanishi, T.; Tokunaga, H.; Susumu, N.; et al. Clinical status and prognostic factors in Japanese patients with uterine leiomyosarcoma. Gynecol. Oncol. 2020, 157, 115–120. [Google Scholar] [CrossRef]
- Look, K.Y.; Sandler, A.; Blessing, J.A.; Lucci, J.A.; Rose, P.G. Phase II trial of gemcitabine as second-line chemotherapy of uterine leiomyosarcoma: A Gynecologic Oncology Group (GOG) Study. Gynecol. Oncol. 2004, 92, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Du, Y.; Qin, C.; Liang, H.; Yang, W.; Lin, J.; Ding, M.; Han, J.; Xu, T. Gemcitabine-Resistant Biomarkers in Bladder Cancer are Associated with Tumor-Immune Microenvironment. Front. Cell Dev. Biol. 2022, 9, 809620. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GSE Series | Platform | No. of Samples | uLMS | Normal Myometrium | ULM | UCS |
|---|---|---|---|---|---|---|
| GSE764 | GPL80 | 26 | 9 | 4 | 7 | / |
| GSE36610 | GPL7363 | 22 | 12 | 10 | / | / |
| GSE64763 | GPL571 | 79 | 25 | 29 | 25 | / |
| GSE68312 | GPL6480 | 9 | 3 | 3 | 3 | |
| GSE32507 | GPL6480 | 46 | 8 | / | / | 14 |
| Category | Term | Count | p-Value |
|---|---|---|---|
| GOTERM_BP_DIRECT | GO:0043627~response to estrogen | 5 | 3.53 × 10−5 |
| GOTERM_BP_DIRECT | GO:0043066~negative regulation of the apoptotic process | 9 | 7.88 × 10−5 |
| GOTERM_BP_DIRECT | GO:0007568~aging | 6 | 1.75 × 10−4 |
| GOTERM_BP_DIRECT | GO:0003151~outflow tract morphogenesis | 4 | 2.91 × 10−4 |
| GOTERM_BP_DIRECT | GO:0045944~positive regulation of transcription from RNA polymerase II promoter | 12 | 3.09 × 10−4 |
| GOTERM_CC_DIRECT | GO:0005887~integral component of plasma membrane | 15 | 1.29 × 10−5 |
| GOTERM_CC_DIRECT | GO:0005886~plasma membrane | 28 | 1.16 × 10−4 |
| GOTERM_CC_DIRECT | GO:0009897~external side of plasma membrane | 8 | 2.41 × 10−4 |
| GOTERM_CC_DIRECT | GO:0005576~extracellular region | 16 | 2.77 × 10−4 |
| GOTERM_CC_DIRECT | GO:0070062~extracellular exosome | 15 | 1.39 × 10−3 |
| GOTERM_MF_DIRECT | GO:0003682~chromatin binding | 8 | 3.57 × 10−4 |
| GOTERM_MF_DIRECT | GO:0042802~identical protein binding | 14 | 8.97 × 10−4 |
| GOTERM_MF_DIRECT | GO:0005539~glycosaminoglycan binding | 3 | 2.49 × 10−3 |
| GOTERM_MF_DIRECT | GO:0005319~lipid transporter activity | 3 | 3.31 × 10−3 |
| GOTERM_MF_DIRECT | GO:0005515~protein binding | 46 | 5.18 × 10−3 |
| KEGG_PATHWAY | hsa05202:Transcriptional misregulation in cancer | 8 | 4.08 × 10−5 |
| KEGG_PATHWAY | hsa05200:Pathways in cancer | 10 | 1.01 × 10−2 |
| KEGG_PATHWAY | hsa05215:Prostate cancer | 4 | 1.23 × 10−2 |
| KEGG_PATHWAY | hsa05205:Proteoglycans in cancer | 5 | 1.86 × 10−2 |
| KEGG_PATHWAY | hsa00240:Pyrimidine metabolism | 3 | 3.38 × 10−2 |
| Category | Term | Count | p-Value |
|---|---|---|---|
| GOTERM_BP_DIRECT | GO:0008284~positive regulation of cell proliferation | 26 | 2 × 10−4 |
| GOTERM_BP_DIRECT | GO:0010628~positive regulation of gene expression | 21 | 6 × 10−6 |
| GOTERM_BP_DIRECT | GO:0007568~aging | 12 | 4 × 10−7 |
| GOTERM_BP_DIRECT | GO:0045944~positive regulation of transcription from RNA polymerase II promoter | 28 | 1 × 10−10 |
| GOTERM_BP_DIRECT | GO:0007179~transforming growth factor beta receptor signaling pathway | 9 | 1 × 10−10 |
| GOTERM_CC_DIRECT | GO:0005615~extracellular space | 40 | 1.98 × 10−7 |
| GOTERM_CC_DIRECT | GO:0005576~extracellular region | 38 | 1.55 × 10−10 |
| GOTERM_CC_DIRECT | GO:0005737~cytoplasm | 72 | 2.06 × 10−10 |
| GOTERM_CC_DIRECT | GO:0031093~platelet alpha granule lumen | 7 | 1.31 × 10−11 |
| GOTERM_CC_DIRECT | GO:0042383~sarcolemma | 8 | 1.53 × 10−11 |
| GOTERM_MF_DIRECT | GO:0005515~protein binding | 134 | 1.19 × 10−10 |
| GOTERM_MF_DIRECT | GO:0005158~insulin receptor binding | 5 | 3.76 × 10−10 |
| GOTERM_MF_DIRECT | GO:0005178~integrin binding | 9 | 5.80 × 10−10 |
| GOTERM_MF_DIRECT | GO:0005509~calcium ion binding | 19 | 6.29 × 10−10 |
| GOTERM_MF_DIRECT | GO:0005114~type II transforming growth factor beta receptor binding | 4 | 6.70 × 10−10 |
| KEGG_PATHWAY | hsa05205:Proteoglycans in cancer | 15 | 5.27 × 10−8 |
| KEGG_PATHWAY | hsa05200:Pathways in cancer | 23 | 1.60 × 10−10 |
| KEGG_PATHWAY | hsa05202:Transcriptional misregulation in cancer | 14 | 1.62 × 10−10 |
| KEGG_PATHWAY | hsa05206:MicroRNAs in cancer | 15 | 6.25 × 10−10 |
| KEGG_PATHWAY | hsa05218:Melanoma | 7 | 3.86 × 10−12 |
| Category | Term | Count | p-Value |
|---|---|---|---|
| GOTERM_BP_DIRECT | GO:0097190~apoptotic signaling pathway | 9 | 1.51 × 10−4 |
| GOTERM_BP_DIRECT | GO:0043086~negative regulation of catalytic activity | 10 | 9.65 × 10−4 |
| GOTERM_BP_DIRECT | GO:0045214~sarcomere organization | 6 | 1.09 × 10−3 |
| GOTERM_BP_DIRECT | GO:0001933~negative regulation of protein phosphorylation | 8 | 1.48 × 10−3 |
| GOTERM_BP_DIRECT | GO:0051893~regulation of focal adhesion assembly | 5 | 1.50 × 10−3 |
| GOTERM_CC_DIRECT | GO:0005925~focal adhesion | 30 | 1.24 × 10−8 |
| GOTERM_CC_DIRECT | GO:0005829~cytosol | 150 | 2.57 × 10−11 |
| GOTERM_CC_DIRECT | GO:0070062~extracellular exosome | 72 | 5.57 × 10−10 |
| GOTERM_CC_DIRECT | GO:0005938~cell cortex | 13 | 1.52 × 10−11 |
| GOTERM_CC_DIRECT | GO:0016020~membrane | 106 | 2.67 × 10−11 |
| GOTERM_MF_DIRECT | GO:0005515~protein binding | 327 | 1.28 × 10−5 |
| GOTERM_MF_DIRECT | GO:0003779~actin binding | 19 | 3.84 × 10−11 |
| GOTERM_MF_DIRECT | GO:0002020~protease binding | 10 | 6.18 × 10−11 |
| GOTERM_MF_DIRECT | GO:0019901~protein kinase binding | 23 | 1.00 × 10−3 |
| GOTERM_MF_DIRECT | GO:0070513~death domain binding | 3 | 4.00 × 10−3 |
| KEGG_PATHWAY | hsa04510:Focal adhesion | 19 | 4.01 × 10−7 |
| KEGG_PATHWAY | hsa04810:Regulation of actin cytoskeleton | 15 | 5.22 × 10−4 |
| KEGG_PATHWAY | hsa04270:Vascular smooth muscle contraction | 11 | 7.31 × 10−4 |
| KEGG_PATHWAY | hsa05135:Yersinia infection | 10 | 3.22 × 10−3 |
| KEGG_PATHWAY | hsa05418:Fluid shear stress and atherosclerosis | 10 | 3.55 × 10−3 |
| UPREGULATED | Category | Term | Count | p-Value |
| GOTERM_BP_DIRECT | GO:0051726~regulation of cell cycle | 3 | 5.02 × 10−3 | |
| GOTERM_BP_DIRECT | GO:0044772~mitotic cell cycle phase transition | 2 | 9.88 × 10−3 | |
| GOTERM_BP_DIRECT | GO:0071897~DNA biosynthetic process | 2 | 1.36 × 10−2 | |
| GOTERM_CC_DIRECT | GO:0000307~cyclin-dependent protein kinase holoenzyme complex | 2 | 1.66 × 10−2 | |
| GOTERM_MF_DIRECT | GO:0019901~protein kinase binding | 3 | 1.87 × 10−2 | |
| KEGG_PATHWAY | hsa00240:Pyrimidine metabolism | 2 | 4.17 × 10−2 | |
| KEGG_PATHWAY | hsa05200:Pathways in cancer | 3 | 5.27 × 10−2 | |
| KEGG_PATHWAY | hsa01232:Nucleotide metabolism | 2 | 6.06 × 10−2 | |
| DOWNREGULATED | GOTERM_BP_DIRECT | GO:0007568~aging | 5 | 7.45 × 10−5 |
| GOTERM_BP_DIRECT | GO:0045944~positive regulation of transcription from RNA polymerase II promoter | 9 | 2.9 × 10−4 | |
| GOTERM_BP_DIRECT | GO:0043066~negative regulation of the apoptotic process | 6 | 9.56 × 10−4 | |
| GOTERM_CC_DIRECT | GO:0005886~plasma membrane | 19 | 1.49 × 10−4 | |
| GOTERM_CC_DIRECT | GO:0005887~integral component of plasma membrane | 9 | 1.03 × 10−3 | |
| GOTERM_CC_DIRECT | GO:0005576~extracellular region | 10 | 3.14 × 10−3 | |
| GOTERM_MF_DIRECT | GO:0005539~glycosaminoglycan binding | 3 | 8.25 × 10−4 | |
| GOTERM_MF_DIRECT | GO:0003682~chromatin binding | 5 | 6.81 × 10−3 | |
| GOTERM_MF_DIRECT | GO:0001228~transcriptional activator activity, RNA polymerase II transcription regulatory region sequence-specific binding | 5 | 7.28 × 10−3 | |
| KEGG_PATHWAY | hsa05202:Transcriptional misregulation in cancer | 7 | 8.54 × 10−6 | |
| KEGG_PATHWAY | hsa05200:Pathways in cancer | 7 | 2.18 × 10−3 | |
| KEGG_PATHWAY | hsa04068:FoxO signaling pathway | 3 | 4.75 × 10−2 |
| Gene Symbol | Gene Name | Regulation | Score |
|---|---|---|---|
| ESR1 | estrogen receptor 1 | downregulated | 60 |
| FOXM1 | forkhead box M1 | upregulated | 55 |
| MMP9 | matrix metallopeptidase 9 | upregulated | 55 |
| IGF1 | insulin-like growth factor 1 | downregulated | 52 |
| CTGF | connective tissue growth factor | downregulated | 51 |
| TK1 | thymidine kinase 1 | upregulated | 50 |
| TYMS | thymidylate synthetase | upregulated | 50 |
| CKS2 | cyclin-dependent kinases regulatory subunit 2 | upregulated | 48 |
| CCNE1 | cyclin E1 | upregulated | 30 |
| TGFBR2 | transforming growth factor, beta receptor II | downregulated | 26 |
| Term | Overlap | Adjusted p-Value | Genes |
|---|---|---|---|
| hsa-miR-26b-5p | 7/1872 | 0.0012 | CCNE1; CKS2; IGF1; TK1; FOXM1; TYMS; CTGF |
| hsa-miR-18b-5p | 3/116 | 0.0029 | IGF1; ESR1; CTGF |
| hsa-miR-302a-5p | 3/126 | 0.0029 | CKS2; IGF1; MMP9 |
| hsa-miR-145-5p | 3/238 | 0.015 | ESR1; CTGF; TGFBR2 |
| hsa-miR-18a-5p | 3/262 | 0.017 | ESR1; CTGF; TGFBR2 |
| Key TF | Description | No. of Overlapped Genes | p-Value | q-Value | List of Overlapped Genes |
|---|---|---|---|---|---|
| SP1 | Sp1 transcription factor | 7 | 6.57 × 10−10 | 1.45 × 10−8 | TGFBR2, FOXM1, TYMS, CTGF, MMP9, TK1, ESR1 |
| FLI1 | Friend leukemia virus integration 1 | 3 | 2.16 × 10−7 | 2.28 × 10−6 | TGFBR2, FOXM1, CTGF |
| HDAC2 | Histone deacetylase 2 | 3 | 3.11 × 10−7 | 2.28 × 10−6 | CCNE1, TGFBR2, IGF1 |
| EP300 | E1A binding protein p300 | 3 | 2.93 × 10−6 | 1.36 × 10−5 | IGF1, CCNE1, MMP9 |
| WT1 | Wilms tumor 1 | 3 | 3.09 × 10−6 | 1.36 × 10−5 | IGF1, CCNE1, CTGF |
| NCOR1 | Nuclear receptor corepressor 1 | 2 | 5.3 × 10−6 | 1.94 × 10−5 | IGF1, ESR1 |
| STAT5B | Signal transducer and activator of transcription 5B | 2 | 7.07 × 10−6 | 2.22 × 10−5 | IGF1, ESR1 |
| ETS1 | V-ets erythroblastosis virus E26 oncogene homolog 1 (avian) | 3 | 8.3 × 10−6 | 2.28 × 10−5 | TGFBR2, CTGF, MMP9 |
| TFDP1 | Transcription factor Dp-1 | 2 | 1.14 × 10−5 | 2.77 × 10−5 | CCNE1, TYMS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rakic, A.; Anicic, R.; Rakic, M.; Nejkovic, L. Integrated Bioinformatics Investigation of Novel Biomarkers of Uterine Leiomyosarcoma Diagnosis and Outcome. J. Pers. Med. 2023, 13, 985. https://doi.org/10.3390/jpm13060985
Rakic A, Anicic R, Rakic M, Nejkovic L. Integrated Bioinformatics Investigation of Novel Biomarkers of Uterine Leiomyosarcoma Diagnosis and Outcome. Journal of Personalized Medicine. 2023; 13(6):985. https://doi.org/10.3390/jpm13060985
Chicago/Turabian StyleRakic, Aleksandar, Radomir Anicic, Marija Rakic, and Lazar Nejkovic. 2023. "Integrated Bioinformatics Investigation of Novel Biomarkers of Uterine Leiomyosarcoma Diagnosis and Outcome" Journal of Personalized Medicine 13, no. 6: 985. https://doi.org/10.3390/jpm13060985