
Ophthalmic Manifestations in Fabry Disease: Updated Review
 , , and
, , and
Abstract
:1. Introduction
2. Epidemiology and Genetics
- Males with no α-gal-A activity. More than 40 mutations of α-gal-A (no α-gal-A activity) have been correlated with the classic phenotype [11,14,15,16]. Individuals with this phenotype develop the full spectrum of clinical signs and symptomatology, with symptom onset in childhood. This mutation occurs in approximately 1 in approximately 37,000 to 60,000 males [15].
- Males with non-classic or atypical FD mutations, resulting in different manifestations. Some gene mutations result in partial α-gal-A activity. These patients may experience symptoms in childhood, but they do not manifest the full spectrum of FD. Compared to the classic phenotype, these variants seem to have more cases of midlife cardiac and renal disease [11].
- Female carriers exhibit a variety of manifestations. Although they were once considered carriers only, females are now recognized as being affected with variable penetrance [11]. The phenotypic variation seen in heterozygotes is caused by random X-inactivation, or lyonization, allowing traits of the mutated X chromosome to be expressed to varying degrees [17,18,19,20].
3. Pathophysiology
4. Conjunctival Manifestations
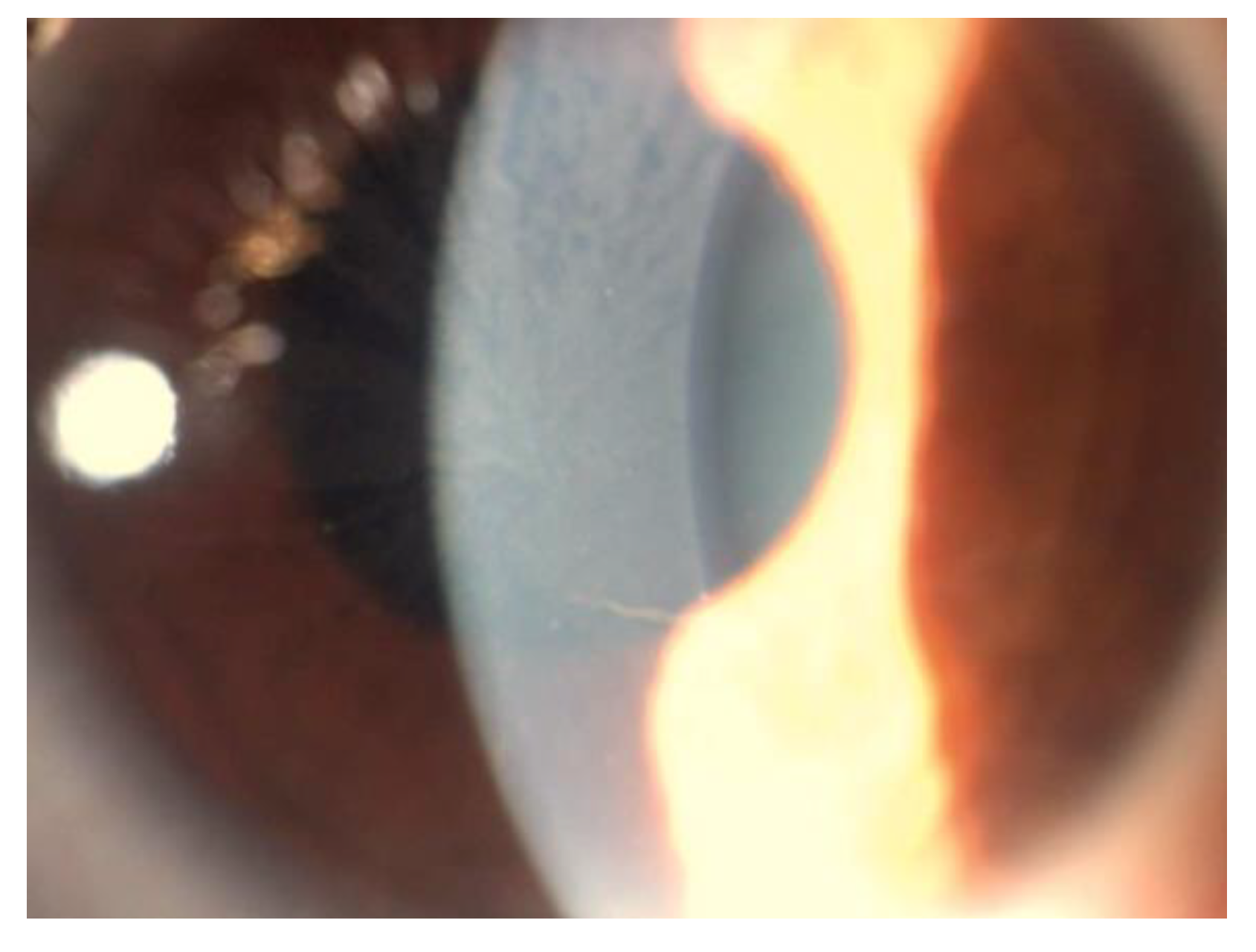
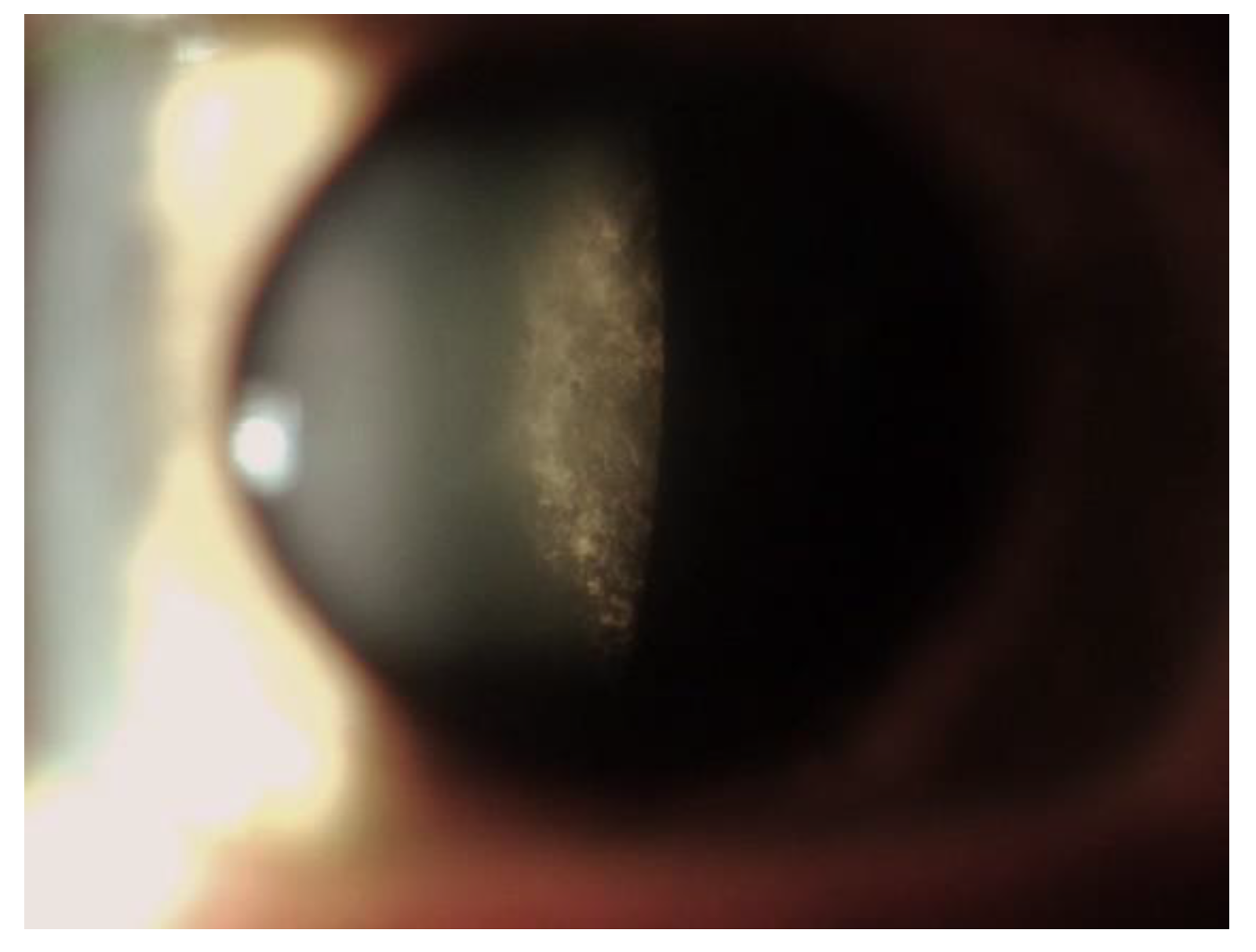
5. Corneal Manifestations
5.1. Histopatological Findings
5.2. Corneal Biomechanics
5.3. In Vivo Confocal Microscopy
5.4. Stromal and Endothelial Disfunction
5.5. Differential Diagnosis of Vortex Keratopathy
6. Lens Manifestations
7. Retinal Manifestations
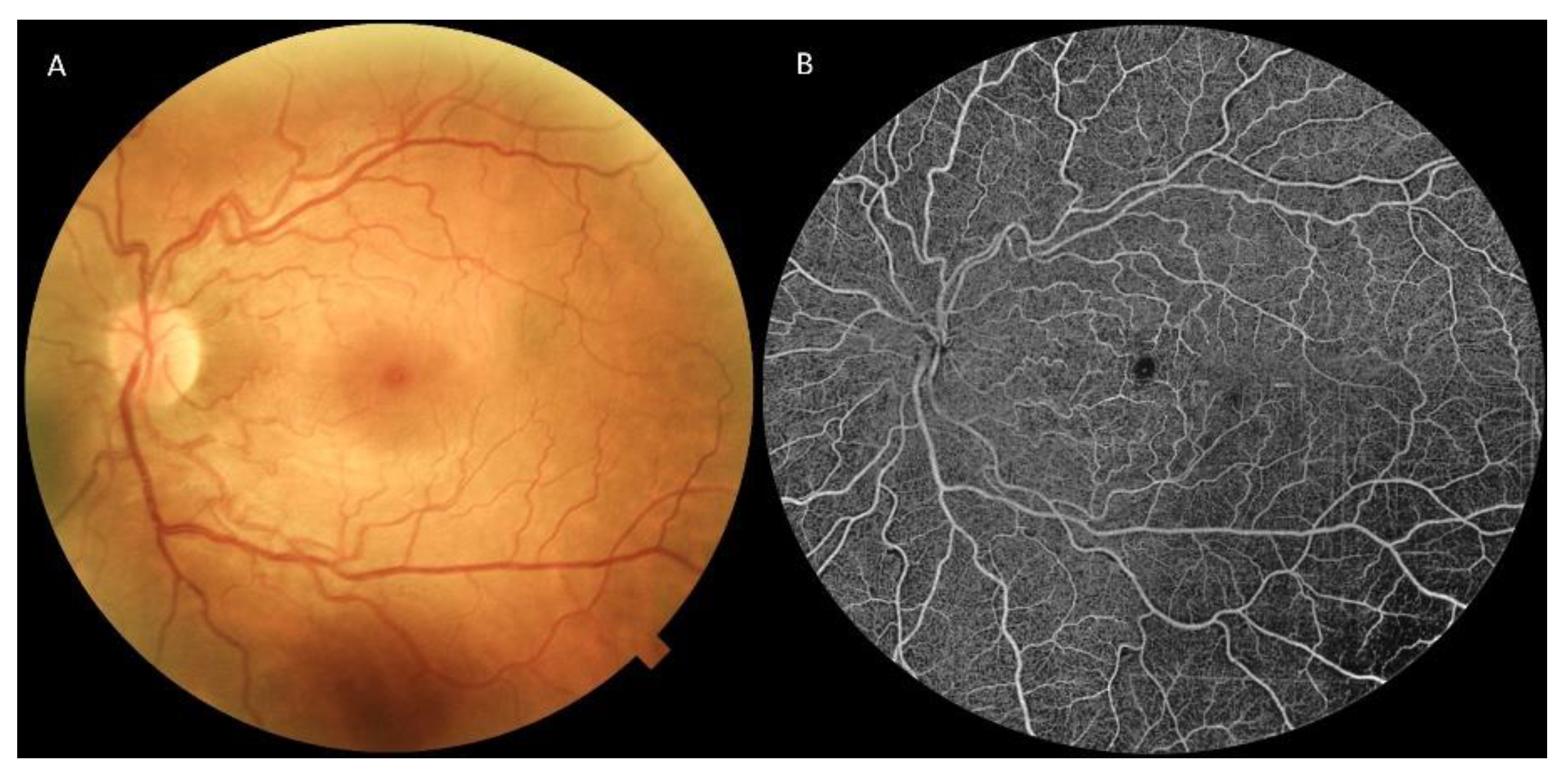
7.1. Retinography
7.2. Fluorescein and Indocyanine Green Angiography
7.3. Optical Coherence Tomography and Optical Coherence Tomography Angiography
7.4. Adaptive Optics
7.5. Electrofunctional Findings
7.6. Correlation with Systemic Manifestations
8. Other Ocular Findings
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- MacDermot, K.D.; Holmes, A.; Miners, A.H. Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J. Med. Genet. 2001, 38, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Sodi, A.; Ioannidis, A.S.; Mehta, A.; Davey, C.; Beck, M.; Pitz, S. Ocular manifestations of Fabry’s disease: Data from the Fabry Outcome Survey. Br. J. Ophthalmol. 2007, 91, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Pitz, S.; Kalkum, G.; Arash, L.; Karabul, N.; Sodi, A.; Larroque, S.; Beck, M.; Gal, A. Ocular signs correlate well with disease severity and genotype in Fabry disease. PLoS ONE 2015, 10, e0120814. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Niemann, M.; Breunig, F.; Herrmann, S.; Beer, M.; Stork, S.; Voelker, W.; Ertl, G.; Wanner, C.; Strotmann, J. Long-term effects of enzyme replacement therapy on fabry cardiomyopathy: Evidence for a better outcome with early treatment. Circulation 2009, 119, 524–529. [Google Scholar] [CrossRef] [PubMed]
- SSpada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High incidence of later-onset fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef]
- Hwu, W.L.; Chien, Y.H.; Lee, N.C.; Chiang, S.C.; Dobrovolny, R.; Huang, A.C.; Yeh, H.-Y.; Chao, M.-C.; Lin, S.-J.; Kitagawa, T.; et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A). Hum. Mutat. 2009, 30, 1397–1405. [Google Scholar] [CrossRef]
- Mehta, A.; Ricci, R.; Widmer, U.; Dehout, F.; de Lorenzo, A.G.; Kampmann, C.; Linhart, A.; Sunder-Plassmann, G.; Ries, M.; Beck, M. Fabry disease defined: Baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur. J. Clin. Investig. 2004, 34, 236–242. [Google Scholar] [CrossRef]
- Mahmud, H.M. Fabry’s disease—A comprehensive review on pathogenesis, diagnosis and treatment. J. Pak. Med. Assoc. 2014, 64, 189–194. [Google Scholar]
- Chan, B.; Adam, D. A Review of Fabry Disease. Ski. Ther. Lett. 2018, 23, 4–6. [Google Scholar]
- Zarate, Y.A.; Hopkin, R.J. Fabry’s disease. Lancet 2008, 372, 1427–1435. [Google Scholar] [CrossRef]
- El-Abassi, R.; Singhal, D.; England, J.D. Fabry’s disease. J. Neurol. Sci. 2014, 344, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, U.; Whybra, C.; Parini, R.; Pintos-Morell, G.; Mehta, A.; Sunder-Plassmann, G.; Widmer, U.; Beck, M.; FOS European Investigators. Clinical manifestations of Fabry disease in children: Data from the Fabry Outcome Survey. Acta Paediatr. 2006, 95, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Bodamer, O.A.; Johnson, B.; Dajnoki, A. Diagnosing lysosomal storage disorders: Fabry disease. Curr. Protoc. Hum. Genet. 2013, 17, 13. [Google Scholar] [CrossRef]
- Ashley, G.A.; Shabbeer, J.; Yasuda, M.; Eng, C.M.; Desnick, R.J. Fabry disease: Twenty novel alpha-galactosidase A mutations causing the classical phenotype. J. Hum. Genet. 2001, 46, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Masson, C.; Cissé, I.; Simon, V.; Insalaco, P.; Audran, M. Fabry disease: A review. Jt. Bone Spine 2004, 71, 381–383. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.F. Fabry Disease: Perspectives from 5 Years of FOS; National Library of Medicine: Bethesda, MD, USA, 2006.
- Lyon, M.F. X-chromosome inactivation and human genetic disease. Acta Paediatr. Suppl. 2002, 91, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolny, R.; Dvorakova, L.; Ledvinova, J.; Magage, S.; Bultas, J.; Lubanda, J.C.; Elleder, M.; Karetova, D.; Pavlikova, M.; Hrebicek, M. Relationship between X-inactivation and clinical involvement in Fabry heterozygotes. Eleven novel mutations in the α-galactosidase A gene in the Czech and Slovak population. J. Mol. Med. 2005, 83, 647–654. [Google Scholar] [CrossRef]
- Maier, E.M.; Osterrieder, S.; Whybra, C.; Ries, M.; Gal, A.; Beck, M.; Roscher, A.A.; Muntau, A.C. Disease manifestations and X inactivation in heterozygous females with Fabry disease. Acta Paediatr. Suppl. 2006, 95, 30–38. [Google Scholar] [CrossRef]
- Germain, D.P. Genetics of Fabry disease: Diagnostic and therapeutic implications. Presse Med. 2007, 36, 1S14-9. [Google Scholar]
- Popli, S.; Leehey, D.J.; Molnar, Z.V.; Nawab, Z.M.; Ing, T.S. Demonstration of Fabry’s disease deposits in placenta. Am. J. Obstet. Gynecol. 1990, 162, 464–465. [Google Scholar] [CrossRef]
- Vedder, A.C.; Strijland, A.; vd Bergh Weerman, M.A.; Florquin, S.; Aerts, J.M.; Hollak, C.E. Manifestations of Fabry disease in placental tissue. J. Inherit. Metab. Dis. 2006, 29, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Pintos-Morell, G.; Beck, M. Fabry disease in children and the effects of enzyme replacement treatment. Eur. J. Pediatr. 2009, 168, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.T.R. Narrative review: Fabry disease. Ann. Intern. Med. 2007, 146, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Kolodny, E.H.; Pastores, G.M. CNS pathology and vascular/circulatory abnormalities in Fabry disease. Acta Paediatr. 2007, 95, 55–56. [Google Scholar] [CrossRef]
- Sodi, A.; Lenzetti, C.; Bacherini, D.; Finocchio, L.; Verdina, T.; Borg, I.; Cipollini, F.; Patwary, F.U.; Tanini, I.; Zoppetti, C.; et al. Quantitative Analysis of Conjunctival and Retinal Vessels in Fabry Disease. J. Ophthalmol. 2019, 2019, 4696429. [Google Scholar] [CrossRef]
- Font, R.L.; Fine, B.S. Ocular pathology in fabry’s disease. Histochemical and electron microscopic observations. Am. J. Ophthalmol. 1972, 73, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Libert, J.; Tondeur, M.; Van Hoff, F. The use of conjunctival biopsy and enzyme analysis in tears for the diagnosis of homozygotes and heterozygotes with Fabry disease. Birth Defects Orig. Artic. Ser. 1976, 12, 221–239. [Google Scholar]
- Libert, J.; Toussaint, D. Tortuosities of retinal and conjunctival vessels in lysosomal storage diseases. Birth Defects Orig. Artic. Ser. 1982, 18, 347–358. [Google Scholar] [PubMed]
- McCulloch, C.; Ghosh, M. Ultrastructural changes in the cornea and conjunctiva of a heterozygous woman with Fabry’s disease. Can. J. Ophthalmol. 1984, 19, 192–198. [Google Scholar]
- Sher, N.A.; Letson, R.D.; Desnick, R.J. The ocular manifestations in Fabry’s disease. Arch. Ophthalmol. 1979, 97, 671–676. [Google Scholar] [CrossRef]
- Cable, W.J.; Kolodny, E.H.; Adams, R.D. Fabry disease: Impaired autonomic function. Neurology 1982, 32, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Hilz, M.J.; Kolodny, E.H.; Brys, M.; Stemper, B.; Haendl, T.; Marthol, H. Reduced cerebral blood flow velocity and impaired cerebral autoregulation in patients with Fabry disease. J. Neurol. 2004, 251, 564–570. [Google Scholar] [CrossRef]
- Orteu, C.; Jansen, T.; Lidove, O.; Jaussaud, R.; Hughes, D.; Pintos-Morell, G.; Ramaswami, U.; Parini, R.; Sunder-Plassman, G.; Beck, M.; et al. Fabry disease and the skin: Data from FOS, the Fabry outcome survey. Br. J. Dermatol. 2007, 157, 331–337. [Google Scholar] [CrossRef]
- Vujkovac, A.C.; Vujkovac, B.; Novakovic, S.; Stevanec, M.; Sabovic, M. Characteristics of Vascular Phenotype in Fabry Patients. Angiology 2021, 72, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Mastropasqua, L.; Nubile, M.; Lanzini, M.; Carpineto, P.; Toto, L.; Ciancaglini, M. Corneal and conjunctival manifestations in Fabry disease: In Vivo confocal microscopy study. Am. J. Ophthalmol. 2006, 141, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Falke, K.; Büttner, A.; Schittkowski, M.; Stachs, O.; Kraak, R.; Zhivov, A.; Rolfs, A.; Guthoff, R. The microstructure of cornea verticillata in Fabry disease and amiodarone-induced keratopathy: A confocal laser-scanning microscopy study. Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.D.; Bower, K.S.; Brooks, D.B.; Walter, A. Fabry disease and chemosis. Cornea 2009, 28, 224–227. [Google Scholar] [CrossRef]
- Sivley, M.D.; Wallace, E.L.; Warnock, D.G.; Benjamin, W.J. Conjunctival lymphangiectasia associated with classic Fabry disease. Br. J. Ophthalmol. 2018, 102, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Ikegawa, Y.; Shiraishi, A.; Hayashi, Y.; Ogimoto, A.; Ohashi, Y. In vivo confocal microscopic observations of vortex keratopathy in patients with amiodarone-induced keratopathy and Fabry disease. J. Ophthalmol. 2018, 2018, 5315137. [Google Scholar] [CrossRef] [PubMed]
- Chierego, C.; Merz, T.; Fasolo, A.; Lagali, N.; Pedrotti, E. In Vivo confocal microscopy of verticillata-like paraproteinemic keratopathy in a patient with monoclonal gammopathy of uncertain significance evolving into smoldering multiple myeloma. Am. J. Ophthalmol. Case Rep. 2019, 15, 100505. [Google Scholar] [CrossRef]
- Fleischer. Über eine eigenartige bisher nicht bekannte Hornhauttrübung (ein Hinweis auf die normale Struktur der Hornhaut?). Albrecht Græfe Arch. Ophthalmol. 1910, 77, 136–140. [Google Scholar] [CrossRef]
- Weicksel, J. Angiomatosis bzw Angiokeratosis universalis. Dtsch. Med. Wochenschr. 1925, 51, 898. [Google Scholar] [CrossRef]
- Franceschetti, A.T. Cornea verticillata (Gruber) and its relation to Fabry’s disease (angiokeratoma corporis diffusum). Ophthalmologica 1968, 156, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Bron, A.J. Vortex patterns of the corneal epithelium. Trans. Ophthalmol. Soc. UK 1973, 93, 455–472. [Google Scholar]
- Dufier, J.L.; Gubler, M.C.; Dhermy, P.; Lenoir, G.; Paupe, J.; Haye, C. Fabry’s disease in ophthalmology (author’s transl). J. Fr. Ophtalmol. 1980, 3, 625–630. [Google Scholar]
- Roussel, T.; Grutzmacher, R.; Coster, D. Patterns of superficial keratopathy. Aust. J. Ophthalmol. 1984, 12, 301–316. [Google Scholar] [CrossRef]
- Moiseev, S.V.; Ismailova, D.S.; Moiseev, A.S.; Bulanov, N.M.; A Karovaikina, E.; Nosova, N.R.; Fomin, V.V. Cornea verticillata in Fabry disease. Ter. Arkhiv 2018, 90, 17–22. [Google Scholar] [CrossRef]
- Rahman, A.N. The ocular manifestations of hereditary dystopic lipidosis (angiokeratoma corporis diffusum universale). Arch. Ophthalmol. 1963, 69, 708–716. [Google Scholar] [CrossRef]
- Orssaud, C.; Dufier, J.; Germain, D. Ocular manifestations in Fabry disease: A survey of 32 hemizygous male patients. Ophthalmic Genet. 2003, 24, 129–139. [Google Scholar] [CrossRef]
- Francois, J.; Hanssens, M.; Teuchy, H. Corneal ultrastructural changes in Fabry’s disease. Ophthalmologica 1977, 179, 7–23. [Google Scholar] [CrossRef]
- Riegel, E.M.; Pokorny, K.S.; Friedman, A.H.; Suhan, J.; Ritch, R.H.; Desnick, R.J. Ocular pathology of Fabry’s disease in a hemizygous male following renal transplantation. Surv. Ophthalmol. 1982, 26, 247–252. [Google Scholar] [CrossRef]
- Tsutsumi, A.; Uchida, Y.; Kanai, T.; Tsutsumi, O.; Satoh, K.; Sakamoto, S. Corneal findings in a foetus with Fabry’s disease. Acta Ophthalmol. 1984, 62, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Macrae, W.G.; Ghosh, M.; McCulloch, C. Corneal changes in Fabry’s disease: A clinico-pathologic case report of a heterozygote. Ophthalmic Paediatr. Genet. 1985, 5, 185–190. [Google Scholar] [CrossRef]
- Hirano, K.; Murata, K.; Miyagawa, A.; Terasaki, H.; Saigusa, J.; Nagasaka, T.; Kobayashi, M. Histopathologic findings of cornea verticillata in a woman heterozygous for Fabry’s disease. Cornea 2001, 20, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Weingeist, T.A.; Blodi, F.C. Fabry’s disease: Ocular findings in a female carrier. A light and electron microscopy study. Arch. Ophthalmol. 1971, 85, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Grace, E.V. Diffuse angiokeratosis (fabry’s disease). Am. J. Ophthalmol. 1966, 62, 139–145. [Google Scholar] [CrossRef]
- Cankurtaran, V.; Tekin, K.; Cakmak, A.I.; Inanc, M.; Turgut, F.H. Assessment of corneal topographic, tomographic, densitometric, and biomechanical properties of Fabry patients with ocular manifestations. Graefe Arch. Clin. Exp. Ophthalmol. 2020, 258, 1057–1064. [Google Scholar] [CrossRef]
- Koh, S.; Haruna, M.; Asonuma, S.; Maeda, N.; Hamano, T.; Sakai, N.; Hara, C.; Maruyama, K.; Nishida, K. Quantitative evaluation of visual function in patients with cornea verticillata associated with Fabry disease. Acta Ophthalmol. 2019, 97, e1098–e1104. [Google Scholar] [CrossRef]
- Leonardi, A.; Carraro, G.; Modugno, R.L.; Rossomando, V.; Scalora, T.; Lazzarini, D.; Calò, L. Cornea verticillata in Fabry disease: A comparative study between slit-lamp examination and in vivo corneal confocal microscopy. Br. J. Ophthalmol. 2020, 104, 718–722. [Google Scholar] [CrossRef]
- Marshall, A.; Tavakoli, M.; Waldek, S.; Efron, N.; Malik, R. Corneal confocal microscopy: A novel noninvasive means to diagnose neuropathy in patients with Fabry disease. Muscle Nerve 2009, 40, 976–984. [Google Scholar]
- Liguori, R.; Di Stasi, V.; Bugiardini, E.; Mignani, R.; Burlina, A.; Borsini, W.; Baruzzi, A.; Montagna, P.; Donadio, V. Small fiber neuropathy in female patients with Fabry disease. Muscle Nerve 2010, 41, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Ries, M. Fabry Disease: A Disorder of Childhood Onset. Pediatr. Neurol. 2016, 64, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Davey, P.G. Fabry disease: A survey of visual and ocular symptoms. Clin. Ophthalmol. 2014, 8, 1555. [Google Scholar] [CrossRef] [PubMed]
- Bitirgen, G.; Turkmen, K.; Malik, R.A.; Ozkagnici, A.; Zengin, N. Corneal confocal microscopy detects corneal nerve damage and increased dendritic cells in Fabry disease. Sci. Rep. 2018, 8, 12244. [Google Scholar] [CrossRef]
- Mayer, W.J.; Mackert, M.J.; Kranebitter, N.; Messmer, E.M.; Grüterich, M.; Kampik, A.; Kook, D. Distribution of antigen presenting cells in the human cornea: Correlation of in vivo confocal microscopy and immunohistochemistry in different pathologic entities. Curr. Eye Res. 2012, 37, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Chong, E.M.; Campbell, R.J.; Bourne, W.M. Vortex keratopathy in a patient with multiple myeloma. Cornea 1997, 16, 592–594. [Google Scholar] [CrossRef]
- Erdurmus, M.; Selcoki, Y.; Yagci, R.; Hepsen, I.F. Amiodarone-induced keratopathy: Full-thickness corneal involvement. Eye Contact Lens. 2008, 34, 131–132. [Google Scholar] [CrossRef]
- Lacava, A.C. Ocular complications of chloroquine and derivatives therapy. Arq. Bras. Oftalmol. 2010, 73, 384–389. [Google Scholar] [CrossRef]
- Wasielica-Poslednik, J.; Pfeiffer, N.; Reinke, J.; Pitz, S. Confocal laser-scanning microscopy allows differentiation between Fabry disease and amiodarone-induced keratopathy. Graefes Arch. Clin. Exp. Ophthalmol. 2011, 249, 1689–1696. [Google Scholar] [CrossRef]
- Ingram, D.; Jaggarao, N.; Chamberlain, D. Ocular changes resulting from therapy with amiodarone. Br. J. Ophthalmol. 1982, 66, 676–679. [Google Scholar] [CrossRef]
- Kaplan, L.J.; Cappaert, W.E. Amiodarone keratopathy: Correlation to dosage and duration. Arch. Ophthalmol. 1982, 100, 601–602. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, K.; Gálvez, J.M.; Gutiérrez, Á.M.; Rico, L.; Criollo, E.; De-la-Torre, A. Ocular findings in Fabry disease in Colombian patients. Biomedica 2019, 39, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Gin, T.; Nicholls, K.; Low, M.; Galanos, J.; Crawford, A. Ophthalmological manifestations of Fabry disease: A survey of patients at the Royal Melbourne Fabry Disease Treatment Centre. Clin. Exp. Ophthalmol. 2005, 33, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Samiy, N. Ocular Features of Fabry Disease: Diagnosis of a Treatable Life-threatening Disorder. Surv. Ophthalmol. 2008, 53, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Sivley, M.D. Fabry disease: A review of ophthalmic and systemic manifestations. Optom. Vis. Sci. 2013, 90, e63–e78. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, H. Several functional and fluorescein fundus angiographic findings in Fabry’s disease. Ophthalmologica 1988, 196, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Kolodny, E.H.; Pastores, G.M. Anderson-Fabry disease: Extrarenal, neurologic manifestations. J. Am. Soc. Nephrol. 2002, 13, S150–S153. [Google Scholar] [CrossRef]
- Moore, D.F.; Kaneski, C.R.; Askari, H.; Schiffmann, R. The cerebral vasculopathy of Fabry disease. J. Neurol. Sci. 2007, 257, 258–263. [Google Scholar] [CrossRef]
- Anastasakis, A.; Papatheodorou, E.; Steriotis, A.K. Fabry disease and cardiovascular involvement. Curr. Pharm. Des. 2013, 19, 5997–6008. [Google Scholar] [CrossRef]
- Abe, H.; Sakai, T.; Sawaguchi, S.; Hasegawa, S.; Takagi, M.; Yoshizawa, T.; Usui, T.; Horikawa, Y. Ischemic optic neuropathy in a female carrier with Fabry’s disease. Ophthalmologica 1992, 205, 83–88. [Google Scholar] [CrossRef]
- Pitz, S.; Grube-Einwald, K.; Renieri, G.; Reinke, J. Subclinical optic neuropathy in Fabry disease. Ophthalmic Genet. 2009, 30, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Sher, N.A.; Reiff, W.; Letson, R.D.; Desnick, R.J. Central retinal artery occlusion complicating Fabry’s disease. Arch. Ophthalmol. 1978, 96, 815–817. [Google Scholar] [CrossRef] [PubMed]
- Dantas, M.A.; Fonseca, R.A.; Kaga, T.; Yannuzzi, L.A.; Spaide, R.F. Retinal and choroidal vascular changes in heterozygous Fabry disease. Retina 2001, 21, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Atiskova, Y.; Rassuli, R.; Koehn, A.F.; Golsari, A.; Wagenfeld, L.; Du Moulin, M.; Muschol, N.; Dulz, S. Retinal hyperreflective foci in Fabry disease. Orphanet J. Rare Dis. 2019, 14, 296. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.; Al-Sheikh, M.; Barthelmes, D.; Nowak, A.; Böni, C.; Zweifel, S.A. Optical Coherence Tomography Angiography findings in patients with Fabry’s disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4249. [Google Scholar]
- Bacherini, D.; Vicini, G.; Nicolosi, C.; Tanini, I.; Lenzetti, C.; Finocchio, L.; Cirami, L.C.; Dervishi, E.; Rizzo, S.; Virgili, G.; et al. Optical Coherence Tomography Angiography for the Evaluation of Retinal Vasculature in Fabry Disease: Our Experience and Review of Current Knowledge. Front. Neurol. 2021, 12, 640719. [Google Scholar] [CrossRef]
- Hufendiek, K.; Kaufeld, J.; Volkmann, I.; Brockmann, D.; Awe, M.; Kromer, R.; Hufendiek, K.; Junker, B.; Framme, C. Density of the Retinal Blood Flow and Thickness Mapping by Spectralis OCT in Patients with Fabry Disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, 5460. [Google Scholar]
- Cakmak, A.I.; Atalay, E.; Cankurtaran, V.; Yasar, E.; Turgut, F.H. Optical coherence tomography angiography analysis of fabry disease. Int. Ophthalmol. 2020, 40, 3023–3032. [Google Scholar] [CrossRef]
- Finocchio, L.; Sodi, A.; Bacherini, D.; Lenzetti, C.; Berni, A.; Mucciolo, D.P.; Tanini, I.; Olivotto, I.; Virgili, G.; Rizzo, S. Optical Coherence Tomography Angiography in Fabry disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2838. [Google Scholar]
- Cennamo, G.; Di Maio, L.G.; Montorio, D.; Tranfa, F.; Russo, C.; Pontillo, G.; Cocozza, S.; Esposito, R.; Di Risi, T.; Imbriaco, M.; et al. Optical Coherence Tomography Angiography Findings in Fabry Disease. J. Clin. Med. 2019, 8, 528. [Google Scholar] [CrossRef]
- Minnella, A.M.; Barbano, L.; Verrecchia, E.; Martelli, F.; Pagliei, V.; Gambini, G.; Placidi, G.; Falsini, B.; Caporossi, A.; Manna, R. Macular Impairment in Fabry Disease: A Morpho-functional Assessment by Swept-Source OCT Angiography and Focal Electroretinography. Invest. Ophthalmol. Vis. Sci. 2019, 60, 2667–2675. [Google Scholar] [CrossRef] [PubMed]
- Dogan, C.; Gonen, B.; Dincer, M.T.; Mergen, B.; Kiykim, E.; Bakir, A.; Trabulus, S.; Yetik, H.; Seyahi, N. Evaluation of the reasons for the microvascular changes in patients with Fabry disease using optic coherence tomography angiography. Eur. J. Ophthalmol. 2021, 31, 3231–3237. [Google Scholar] [CrossRef] [PubMed]
- Sodi, A.; Germain, D.P.; Bacherini, D.; Finocchio, L.; Pacini, B.; Marziali, E.; Lenzetti, C.; Tanini, I.; Koraichi, F.; Coriat, C.; et al. In Vivo observation of retinal vascular deposits using adaptive optics imaging in Fabry disease. Retina 2020, 40, 1623–1629. [Google Scholar] [CrossRef]
- Bitirgen, G.; Turkmen, K.; Zengin, N.; Malik, R.A. Altered pupillary light responses are associated with the severity of autonomic symptoms in patients with Fabry disease. Sci. Rep. 2021, 11, 8146. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Moore, D.F.; Robinson, C.J.; Tifft, C.J.; Rosenbaum, K.N.; Brady, R.O.; Schiffmann, R.; Krasnewich, D. Quantitative dysmorphology assessment in Fabry disease. Genet. Med. 2006, 8, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Cox-Brinkman, J.; Vedder, A.; Hollak, C.; Richfield, L.; Mehta, A.; Orteu, K.; Wijburg, F.; Hammond, P. Three-dimensional face shape in Fabry disease. Eur. J. Hum. Genet. 2007, 15, 535–542. [Google Scholar] [CrossRef]
- Shen, Y.D.; Yang, C.M.; Huang, J.S. Fabry disease manifesting as chronic uveitis—Treated with enzyme replacement therapy. Eye 2007, 21, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Sodi, A.; Bini, A.; Mignani, R.; Minuti, B.; Menchini, U. Subfoveal choroidal neovascularization in a patient with Fabry’s disease. Int. Ophthalmol. 2009, 29, 435–437. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Study Authors | OCTA Devices | Scan Width (mm) | No. of FD/Controls | Findings in FD Patients |
|---|---|---|---|---|
| Finocchio et al. (2018) [90] | Spectral Domain | 3 × 3 | 13/13 | ↓ vascular density in DCP, = SCP; ↓ FAZ of SCP and DCP |
| Hufendiek et al. (2018) [88] | Spectral Domain | 3 × 3 | 10/10 | ↓ flow density in DCP, SCP and choriocapillaris |
| Baur et al. (2018) [86] | Swept Source | 3 × 3 | 14/8 | ↓ vascular density in DCP, = in SCP |
| Cennamo et al. (2019) [91] | Spectral Domain | 6 × 6 | 54/70 | ↓ vascular density in SCP, ↑ in DCP |
| Minnella et al. (2019) [92] | Swept Source | 4.5 × 4.5 | 20/17 | ↑ vascular density in SCP, = in DCP ↑ FAZ in SCP and DCP |
| Cakmak et al. (2020) [89] | Spectral Domain | Macula: 6 × 6 Disk: 4.5 × 4.5 | 25/37 | ↓ vessel density in SCP and DCP; ↑ FAZ of SCP and DCP = density of radial peripapillary capillaries |
| Dogan et al. (2021) [93] | Spectral Domain | 6 × 6 | 38/40 | ↓ vascular density in DCP, = in SCP and choriocapillaris |
| Bacherini et al. (2021) [87] | Spectral Domain | 3 × 3 | 13/13 | ↓ vascular density in SCP and DCP; = vessel perfusion and FAZ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambini, G.; Scartozzi, L.; Giannuzzi, F.; Carlà, M.M.; Boselli, F.; Caporossi, T.; De Vico, U.; Baldascino, A.; Rizzo, S. Ophthalmic Manifestations in Fabry Disease: Updated Review. J. Pers. Med. 2023, 13, 904. https://doi.org/10.3390/jpm13060904
Gambini G, Scartozzi L, Giannuzzi F, Carlà MM, Boselli F, Caporossi T, De Vico U, Baldascino A, Rizzo S. Ophthalmic Manifestations in Fabry Disease: Updated Review. Journal of Personalized Medicine. 2023; 13(6):904. https://doi.org/10.3390/jpm13060904
Chicago/Turabian StyleGambini, Gloria, Luca Scartozzi, Federico Giannuzzi, Matteo Mario Carlà, Francesco Boselli, Tomaso Caporossi, Umberto De Vico, Antonio Baldascino, and Stanislao Rizzo. 2023. "Ophthalmic Manifestations in Fabry Disease: Updated Review" Journal of Personalized Medicine 13, no. 6: 904. https://doi.org/10.3390/jpm13060904