
Novel WDR72 Mutations Causing Hypomaturation Amelogenesis Imperfecta
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Enrollment of the Study Families
2.2. Genomic DNA Isolation and WES
2.3. Bioinformatic Analysis
2.4. Sanger Sequencing
2.5. Exon Deletion Characterization
3. Results
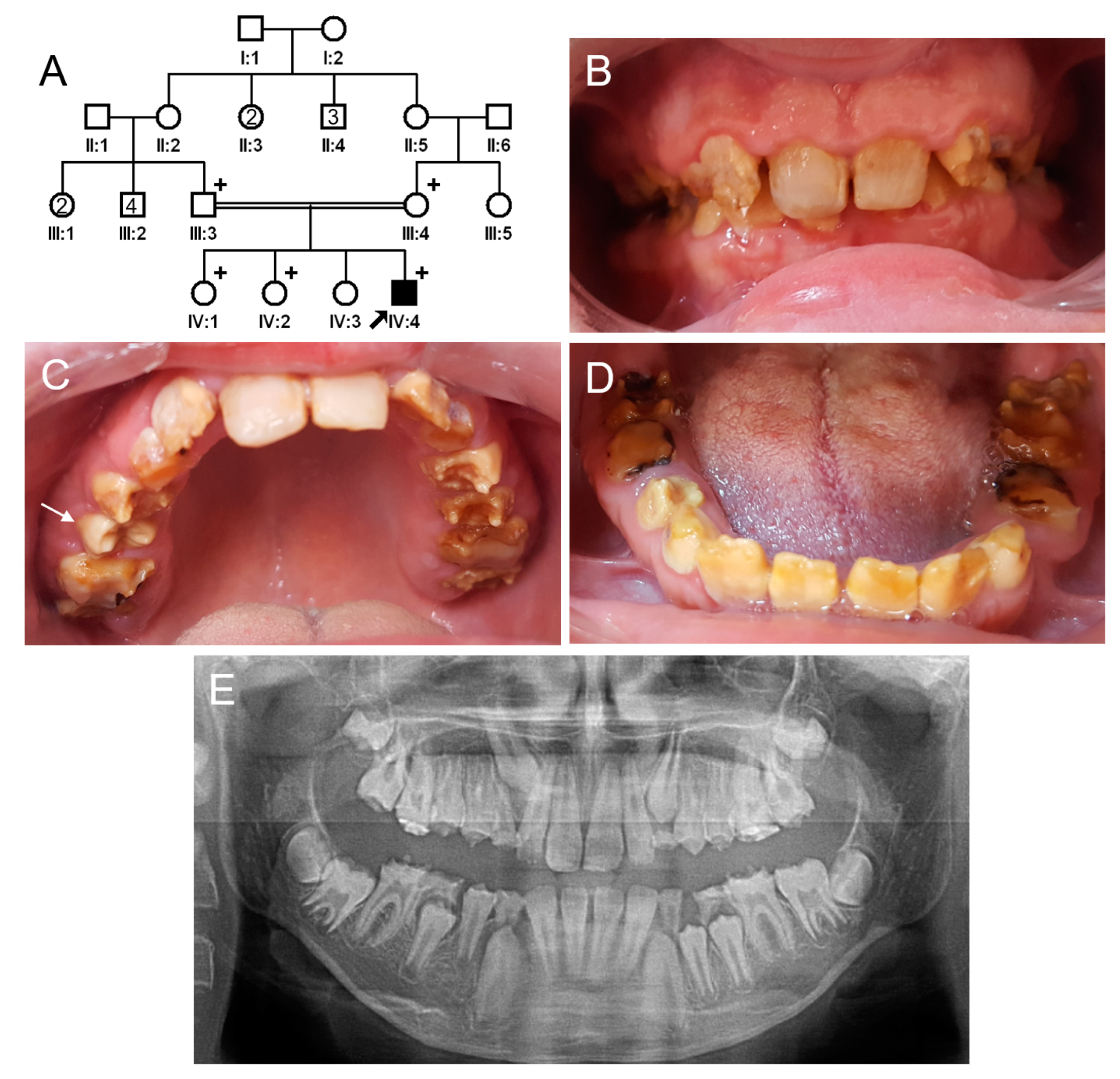
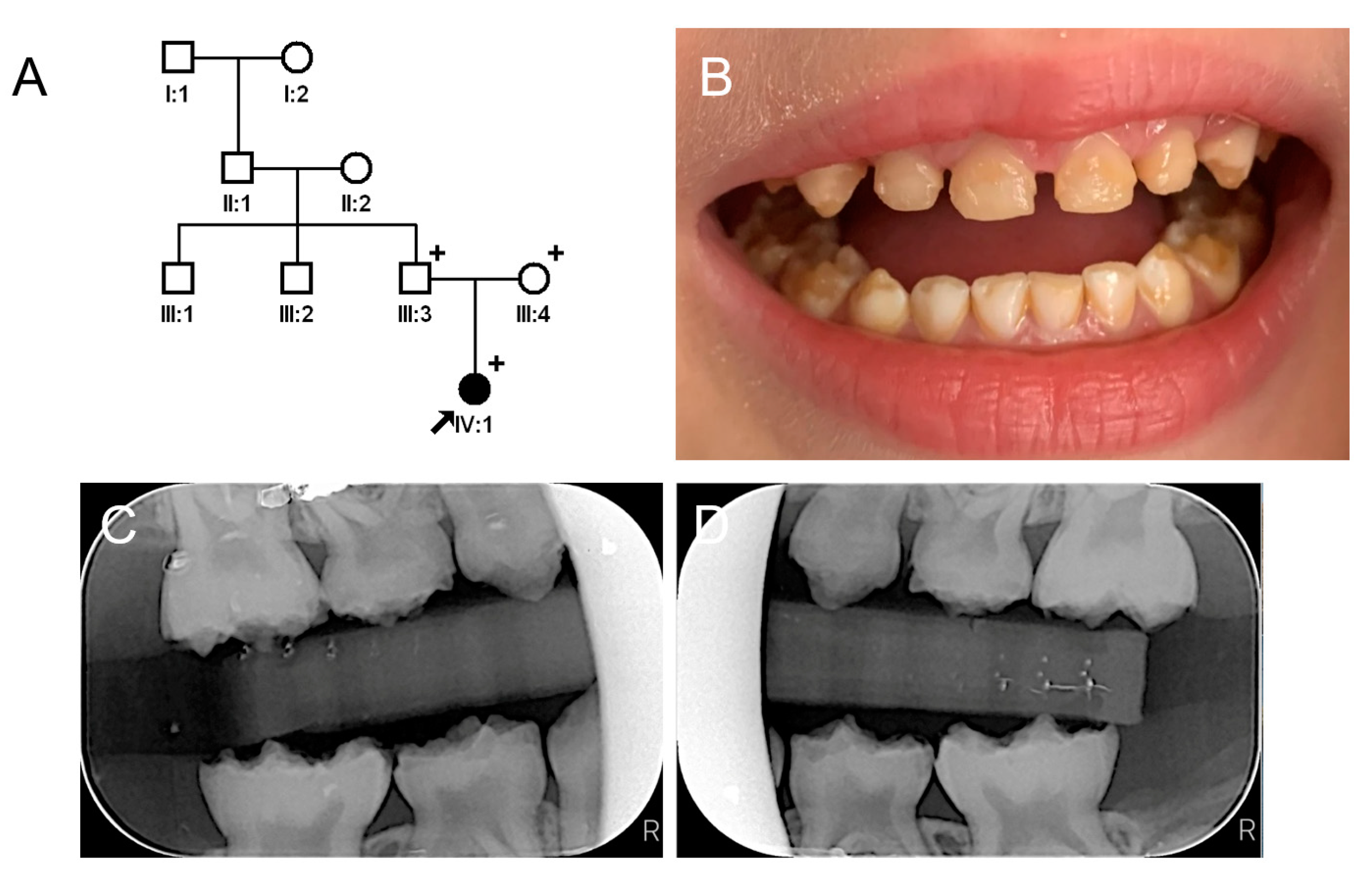
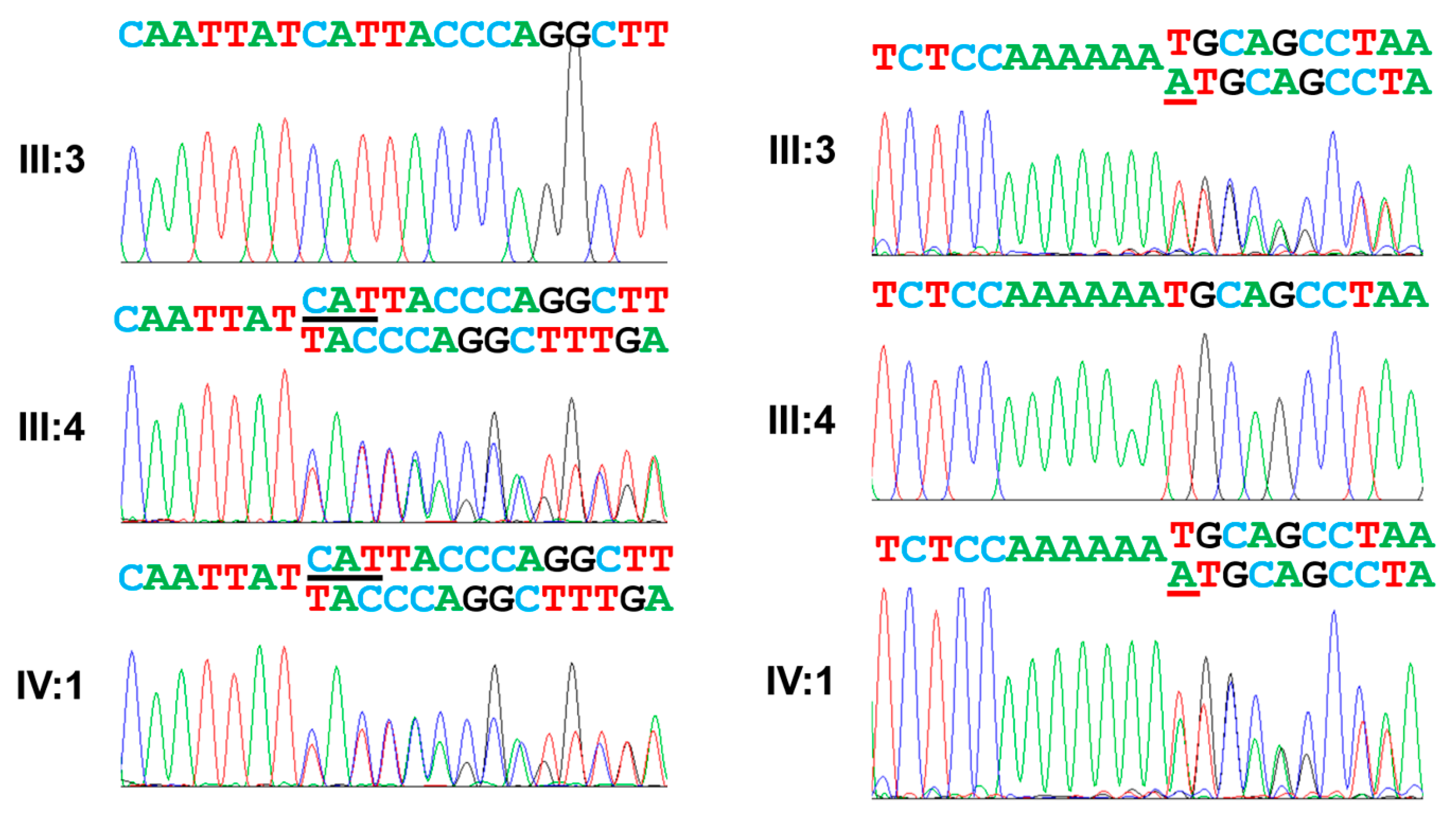
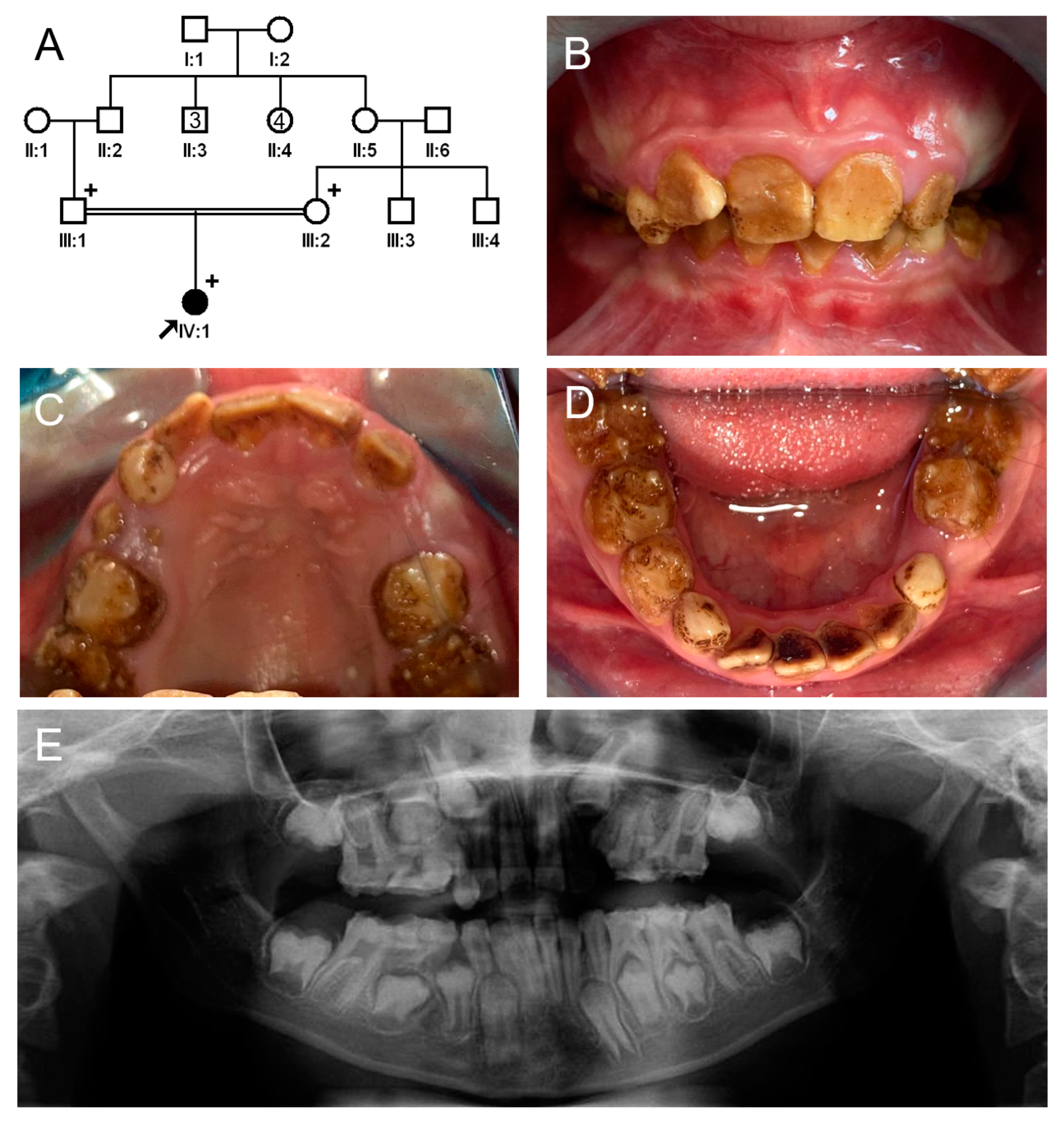
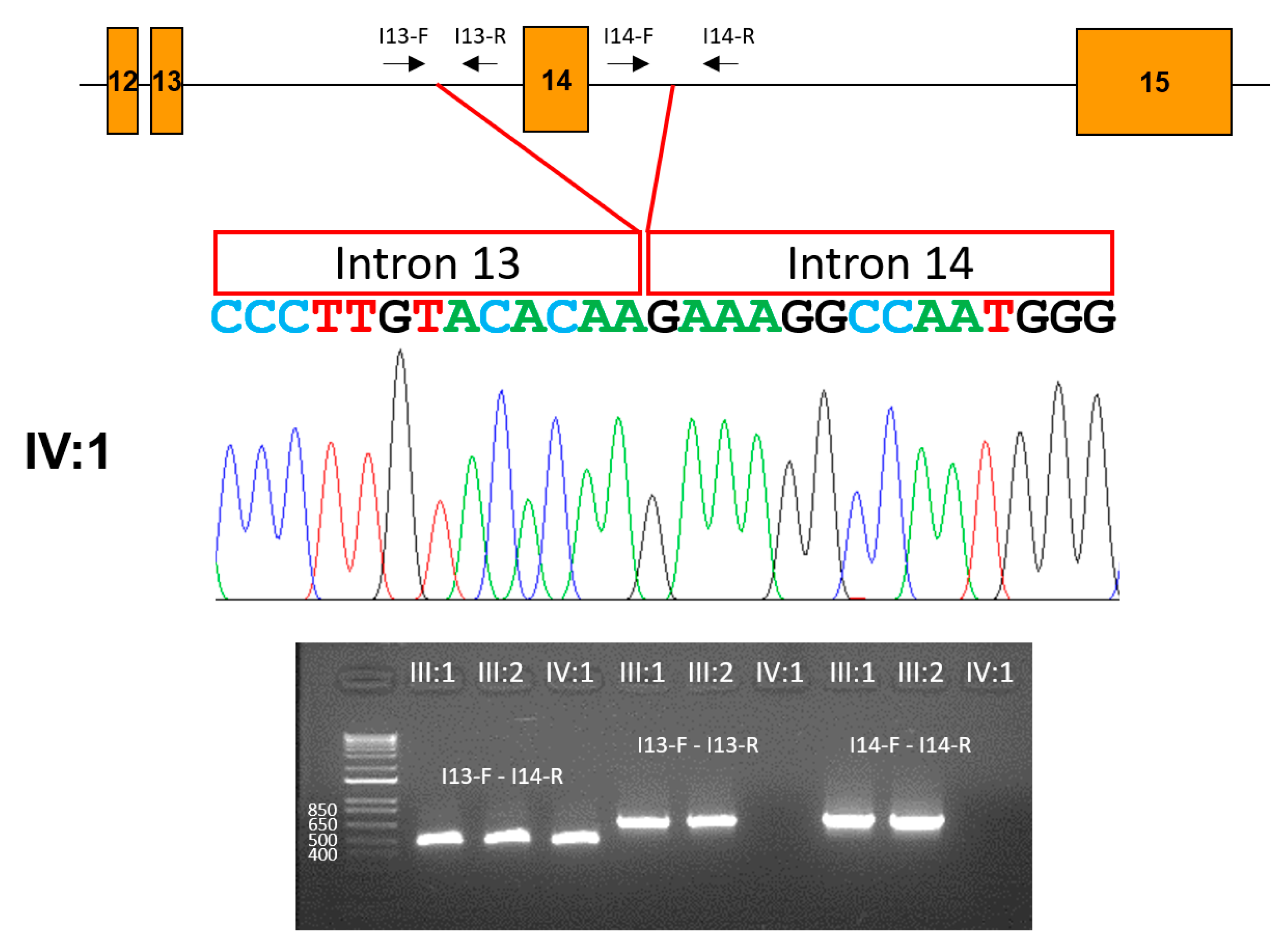
3.1. Family 1
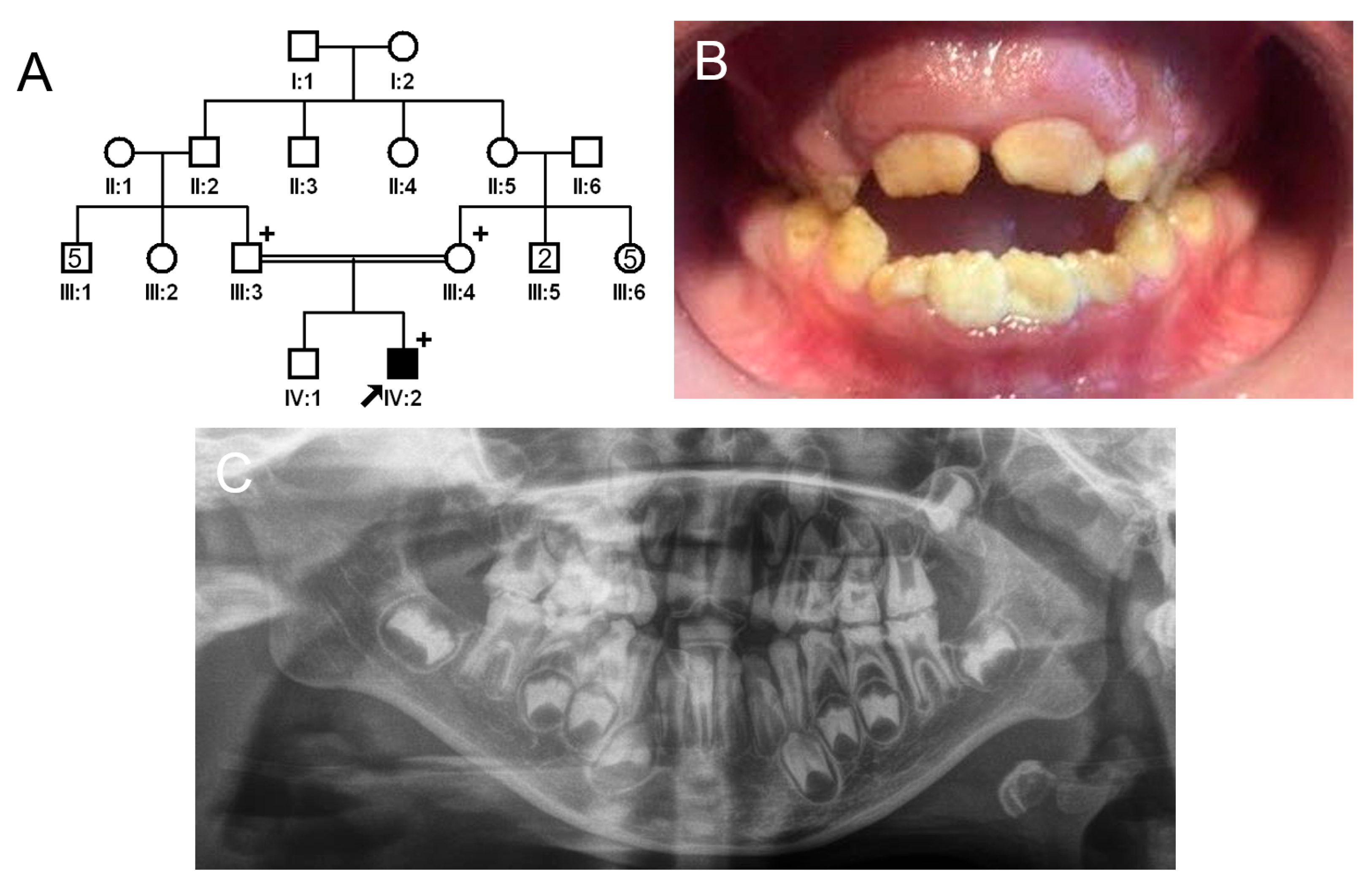
3.2. Family 2
3.3. Family 3
3.4. Family 4
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wright, J.T.; Carrion, I.A.; Morris, C. The molecular basis of hereditary enamel defects in humans. J. Dent. Res. 2015, 94, 52–61. [Google Scholar] [CrossRef]
- Witkop, C.J., Jr. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: Problems in classification. J. Oral Pathol. 1988, 17, 547–553. [Google Scholar] [CrossRef]
- Wright, J.T.; Hong, S.P.; Simmons, D.; Daly, B.; Uebelhart, D.; Luder, H.U. DLX3 c.561_562delCT mutation causes attenuated phenotype of tricho-dento-osseous syndrome. Am. J. Med. Genet. A 2008, 146, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Price, J.A.; Wright, J.T.; Walker, S.J.; Crawford, P.J.; Aldred, M.J.; Hart, T.C. Tricho-dento-osseous syndrome and amelogenesis imperfecta with taurodontism are genetically distinct conditions. Clin. Genet. 1999, 56, 35–40. [Google Scholar] [CrossRef]
- O’Sullivan, J.; Bitu, C.C.; Daly, S.B.; Urquhart, J.E.; Barron, M.J.; Bhaskar, S.S.; Martelli-Junior, H.; dos Santos Neto, P.E.; Mansilla, M.A.; Murray, J.C.; et al. Whole-Exome sequencing identifies FAM20A mutations as a cause of amelogenesis imperfecta and gingival hyperplasia syndrome. Am. J. Hum. Genet. 2011, 88, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.K.; Reid, B.M.; Dugan, S.L.; Roggenbuck, J.A.; Read, L.; Aref, P.; Taheri, A.P.; Yeganeh, M.Z.; Simmer, J.P.; Hu, J.C. FAM20A mutations associated with enamel renal syndrome. J. Dent. Res. 2014, 93, 42–48. [Google Scholar] [CrossRef]
- Folayan, M.O.; El Tantawi, M.; Oginni, A.B.; Alade, M.; Adeniyi, A.; Finlayson, T.L. Malnutrition, enamel defects, and early childhood caries in preschool children in a sub-urban Nigeria population. PLoS ONE 2020, 15, e0232998. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.C.; Estrella, N.M.; Milkovich, R.N.; Kim, J.W.; Simmer, J.P.; Hu, J.C. Target gene analyses of 39 amelogenesis imperfecta kindreds. Eur. J. Oral Sci. 2011, 119 (Suppl. 1), 311–323. [Google Scholar] [CrossRef]
- Kim, J.W.; Zhang, H.; Seymen, F.; Koruyucu, M.; Hu, Y.; Kang, J.; Kim, Y.J.; Ikeda, A.; Kasimoglu, Y.; Bayram, M.; et al. Mutations in RELT cause autosomal recessive amelogenesis imperfecta. Clin. Genet. 2019, 95, 375–383. [Google Scholar] [CrossRef]
- Cho, S.H.; Seymen, F.; Lee, K.E.; Lee, S.K.; Kweon, Y.S.; Kim, K.J.; Jung, S.E.; Song, S.J.; Yildirim, M.; Bayram, M.; et al. Novel FAM20A mutations in hypoplastic amelogenesis imperfecta. Hum. Mutat. 2012, 33, 91–94. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Kaewgahya, M.; Khemaleelakul, U.; Dejkhamron, P.; Sutthimethakorn, S.; Thongboonkerd, V.; Iamaroon, A. Enamel-renal-gingival syndrome and FAM20A mutations. Am. J. Med. Genet. A 2014, 164A, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Seymen, F.; Lin, B.P.; Kiziltan, B.; Gencay, K.; Simmer, J.P.; Hu, J.C. ENAM mutations in autosomal-dominant amelogenesis imperfecta. J. Dent. Res. 2005, 84, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, D.; Hart, P.S.; Firatli, E.; Aren, G.; Ryu, O.H.; Hart, T.C. Phenotype of ENAM mutations is dosage-dependent. J. Dent. Res. 2005, 84, 1036–1041. [Google Scholar] [CrossRef]
- Wright, J.T.; Hart, P.S.; Aldred, M.J.; Seow, K.; Crawford, P.J.; Hong, S.P.; Gibson, C.W.; Hart, T.C. Relationship of phenotype and genotype in X-linked amelogenesis imperfecta. Connect. Tissue Res. 2003, 44 (Suppl. 1), 72–78. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.K.; Laouina, S.; El Alloussi, M.; Dollfus, H.; Bloch-Zupan, A. Amelogenesis Imperfecta: 1 Family, 2 Phenotypes, and 2 Mutated Genes. J. Dent. Res. 2016, 95, 1457–1463. [Google Scholar] [CrossRef]
- Kim, J.W.; Simmer, J.P.; Hart, T.C.; Hart, P.S.; Ramaswami, M.D.; Bartlett, J.D.; Hu, J.C. MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J. Med. Genet. 2005, 42, 271–275. [Google Scholar] [CrossRef]
- Hart, P.S.; Hart, T.C.; Michalec, M.D.; Ryu, O.H.; Simmons, D.; Hong, S.; Wright, J.T. Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J. Med. Genet. 2004, 41, 545–549. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, W.; Parry, D.A.; Shore, R.C.; Ahmed, M.; Jafri, H.; Rashid, Y.; Al-Bahlani, S.; Al Harasi, S.; Kirkham, J.; Inglehearn, C.F.; et al. Mutations in the beta propeller WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta. Am. J. Hum. Genet. 2009, 85, 699–705. [Google Scholar] [CrossRef]
- Parry, D.A.; Brookes, S.J.; Logan, C.V.; Poulter, J.A.; El-Sayed, W.; Al-Bahlani, S.; Al Harasi, S.; Sayed, J.; Raifel, M.; Shore, R.C.; et al. Mutations in C4orf26, encoding a peptide with in vitro hydroxyapatite crystal nucleation and growth activity, cause amelogenesis imperfecta. Am. J. Hum. Genet. 2012, 91, 565–571. [Google Scholar] [CrossRef]
- Parry, D.A.; Poulter, J.A.; Logan, C.V.; Brookes, S.J.; Jafri, H.; Ferguson, C.H.; Anwari, B.M.; Rashid, Y.; Zhao, H.; Johnson, C.A.; et al. Identification of mutations in SLC24A4, encoding a potassium-dependent sodium/calcium exchanger, as a cause of amelogenesis imperfecta. Am. J. Hum. Genet. 2013, 92, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Parry, D.A.; Smith, C.E.; El-Sayed, W.; Poulter, J.A.; Shore, R.C.; Logan, C.V.; Mogi, C.; Sato, K.; Okajima, F.; Harada, A.; et al. Mutations in the pH-Sensing G-protein-Coupled Receptor GPR68 Cause Amelogenesis Imperfecta. Am. J. Hum. Genet. 2016, 99, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Price, J.A.; Bowden, D.W.; Wright, J.T.; Pettenati, M.J.; Hart, T.C. Identification of a mutation in DLX3 associated with tricho-dento-osseous (TDO) syndrome. Hum. Mol. Genet. 1998, 7, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Hyun, H.K.; Kim, J.W. Thickness and microhardness of deciduous tooth enamel with known DLX3 mutation. Arch. Oral Biol. 2009, 54, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Ravassipour, D.B.; Hart, P.S.; Hart, T.C.; Ritter, A.V.; Yamauchi, M.; Gibson, C.; Wright, J.T. Unique enamel phenotype associated with amelogenin gene (AMELX) codon 41 point mutation. J. Dent. Res. 2000, 79, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Seymen, F.; Lee, K.E.; Kang, H.Y.; Yildirim, M.; Tuna, E.B.; Gencay, K.; Hwang, Y.H.; Nam, K.H.; De La Garza, R.J.; et al. Novel WDR72 mutation and cytoplasmic localization. J. Dent. Res. 2010, 89, 1378–1382. [Google Scholar] [CrossRef]
- Kim, Y.J.; Lee, Y.; Kasimoglu, Y.; Seymen, F.; Simmer, J.P.; Hu, J.C.; Cho, E.S.; Kim, J.W. Recessive Mutations in ACP4 Cause Amelogenesis Imperfecta. J. Dent. Res. 2022, 101, 37–45. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Wright, J.T.; Torain, M.; Long, K.; Seow, K.; Crawford, P.; Aldred, M.J.; Hart, P.S.; Hart, T.C. Amelogenesis imperfecta: Genotype-phenotype studies in 71 families. Cells Tissues Organs 2011, 194, 279–283. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Koruyucu, M.; Seymen, F.; Kasimoglu, Y.; Kim, J.W.; Tinawi, S.; Zhang, C.; Jacquemont, M.L.; Vieira, A.R.; Simmer, J.P.; et al. WDR72 Mutations Associated with Amelogenesis Imperfecta and Acidosis. J. Dent. Res. 2019, 98, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Neer, E.J.; Schmidt, C.J.; Nambudripad, R.; Smith, T.F. The ancient regulatory-protein family of WD-repeat proteins. Nature 1994, 371, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Jain, B.P.; Pandey, S. WD40 Repeat Proteins: Signalling Scaffold with Diverse Functions. Protein J. 2018, 37, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.F. Diversity of WD-repeat proteins. Subcell. Biochem. 2008, 48, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.A.; Ponting, C.P.; Gibson, T.J.; Bork, P. Homology-based method for identification of protein repeats using statistical significance estimates. J. Mol. Biol. 2000, 298, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef]
- Katsura, K.A.; Horst, J.A.; Chandra, D.; Le, T.Q.; Nakano, Y.; Zhang, Y.; Horst, O.V.; Zhu, L.; Le, M.H.; DenBesten, P.K. WDR72 models of structure and function: A stage-specific regulator of enamel mineralization. Matrix Biol. 2014, 38, 48–58. [Google Scholar] [CrossRef]
- Köttgen, A.; Pattaro, C.; Böger, C.A.; Fuchsberger, C.; Olden, M.; Glazer, N.L.; Parsa, A.; Gao, X.; Yang, Q.; Smith, A.V.; et al. New loci associated with kidney function and chronic kidney disease. Nat. Genet. 2010, 42, 376–384. [Google Scholar] [CrossRef]
- Gorski, M.; Jung, B.; Li, Y.; Matias-Garcia, P.R.; Wuttke, M.; Coassin, S.; Thio, C.H.L.; Kleber, M.E.; Winkler, T.W.; Wanner, V.; et al. Meta-analysis uncovers genome-wide significant variants for rapid kidney function decline. Kidney Int. 2021, 99, 926–939. [Google Scholar] [CrossRef]
- Rungroj, N.; Nettuwakul, C.; Sawasdee, N.; Sangnual, S.; Deejai, N.; Misgar, R.A.; Pasena, A.; Khositseth, S.; Kirdpon, S.; Sritippayawan, S.; et al. Distal renal tubular acidosis caused by tryptophan-aspartate repeat domain 72 (WDR72) mutations. Clin. Genet. 2018, 94, 409–418. [Google Scholar] [CrossRef]
- Khandelwal, P.; Mahesh, V.; Mathur, V.P.; Raut, S.; Geetha, T.S.; Nair, S.; Hari, P.; Sinha, A.; Bagga, A. Phenotypic variability in distal acidification defects associated with WDR72 mutations. Pediatr. Nephrol. 2021, 36, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Jobst-Schwan, T.; Klämbt, V.; Tarsio, M.; Heneghan, J.F.; Majmundar, A.J.; Shril, S.; Buerger, F.; Ottlewski, I.; Shmukler, B.E.; Topaloglu, R.; et al. Whole exome sequencing identified ATP6V1C2 as a novel candidate gene for recessive distal renal tubular acidosis. Kidney Int. 2020, 97, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Kuechler, A.; Hentschel, J.; Kurth, I.; Stephan, B.; Prott, E.C.; Schweiger, B.; Schuster, A.; Wieczorek, D.; Ludecke, H.J. A Novel Homozygous WDR72 Mutation in Two Siblings with Amelogenesis Imperfecta and Mild Short Stature. Mol. Syndromol. 2012, 3, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Hentschel, J.; Tatun, D.; Parkhomchuk, D.; Kurth, I.; Schimmel, B.; Heinrich-Weltzien, R.; Bertzbach, S.; Peters, H.; Beetz, C. Identification of the first multi-exonic WDR72 deletion in isolated amelogenesis imperfecta, and generation of a WDR72-specific copy number screening tool. Gene 2016, 590, 1–4. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, W.; Shore, R.C.; Parry, D.A.; Inglehearn, C.F.; Mighell, A.J. Hypomaturation amelogenesis imperfecta due to WDR72 mutations: A novel mutation and ultrastructural analyses of deciduous teeth. Cells Tissues Organs 2011, 194, 60–66. [Google Scholar] [CrossRef]
- Katsura, K.; Nakano, Y.; Zhang, Y.; Shemirani, R.; Li, W.; Den Besten, P. WDR72 regulates vesicle trafficking in ameloblasts. Sci. Rep. 2022, 12, 2820. [Google Scholar] [CrossRef]
- Husein, D.; Alamoudi, A.; Ohyama, Y.; Mochida, H.; Ritter, B.; Mochida, Y. Identification of the C-terminal region in Amelogenesis Imperfecta causative protein WDR72 required for Golgi localization. Sci. Rep. 2022, 12, 4640. [Google Scholar] [CrossRef]
- Ouyang, X.; Shi, X.; Huang, N.; Yang, Y.; Zhao, W.; Guo, W.; Huang, Y. WDR72 Enhances the Stemness of Lung Cancer Cells by Activating the AKT/HIF-1α Signaling Pathway. J. Oncol. 2022, 2022, 5059588. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.K.; Hu, Y.; Yang, J.; Smith, C.E.; Nunez, S.M.; Richardson, A.S.; Pal, S.; Samann, A.C.; Hu, J.C.; Simmer, J.P. Critical roles for WDR72 in calcium transport and matrix protein removal during enamel maturation. Mol. Genet. Genom. Med. 2015, 3, 302–319. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Chromosomal Location | Number of Exon | Length of Protein (aa) | OMIM |
|---|---|---|---|---|
| MMP20 | 11q22.2 | 10 (NM_004771.4) | 483 (NP_004762.2) | *604629 |
| KLK4 | 19q13.41 | 6 (NM_004917.5) | 254 (NP_004908.4) | *603767 |
| WDR72 | 15q21.3 | 20 (NM_182758.4) | 1102 (NP_877435.3) | *613214 |
| ODAPH | 4q21.1 | 2 (NM_178497.5) | 130 (NP_848592.2) | *614829 |
| SLC24A4 | 14q32.12 | 17 (NM_153646.4) | 622 (NP_705932.2) | *609840 |
| GPR68 | 14q32.11 | 2 (NM_001177676.2) | 365 (NP_001171147.1) | *601404 |
| Location | cDNA | Protein | Mode of Inheritance | References |
|---|---|---|---|---|
| Exon 2 | c.88C>T | p.(Arg30*) | Homozygous | Khandelwal et al. (2021) [41] |
| Exon 3 | c.154_1765del (exon 3–13 del) | p.(Ile52Alafs*25) | Homozygous | Zhang et al. (2019) [31] |
| Exon 5 | c.477_485dup | p.(Ile159_Cys161dup) | Homozygous | Jobst-Schwan et al. (2020) [42] |
| Exon 5 | c.377G>A | p.(Trp126*) | Homozygous | Zhang et al. (2019) [31] |
| Exon 8 | c.764_768delGGCAG | p.(Gly255Valfs*40) | Homozygous | Jobst-Schwan et al. (2020) [42] |
| Exon 8 | c.806_810delGGCAG | p.(Gly255Valfs*294) | Homozygous | Katsura et al. (2014) [37] |
| Exon 10 | c.997A>T | p.(Lys333*) | Homozygous | Kuechler et al. (2012) [43] |
| Exon 11 | c.1265G>T | p.(Gly422Val) | Homozygous | Zhang et al. (2019) [31] |
| Exon 11 | c.1287_1289delCAT | p.(Ile430del) | Maternal | This report |
| Exon 12 | c.1467_1468delAT | p.(Val491Aspfs*8) | Homozygous | Lee et al. (2010) [25] Wright et al. (2011) [30] Zhang et al. (2019) [31] This report |
| Exon 12 | c.1481G>A | p.(Trp494*) | Homozygous | Khandelwal et al. (2021) [41] |
| Exon 13 | c.1570_3148del (exon 13–18 del) | p.(Arg525Glnfs*43) | Homozygous | Hentschel et al. (2016) [44] |
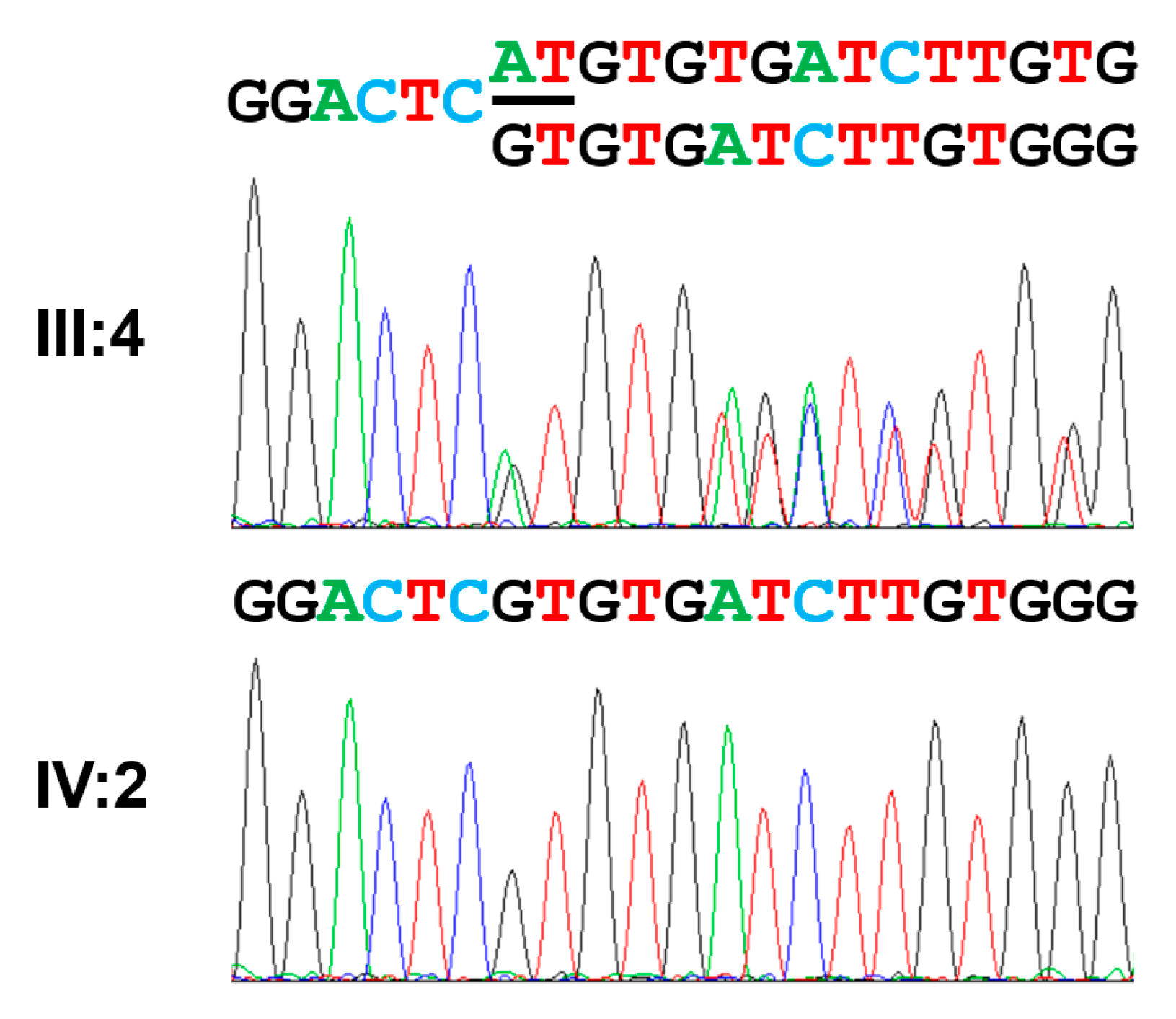
| Exon 14 | c.1766_1964del (exon 14 del) | p.(Gly589Valfs*16) | Homozygous | This report |
| Exon 14 | c.1777A>G | p.(Arg593Gly) | Paternal | Rungroj et al. (2018) [40] |
| Exon 14 | c.1801C>T | p.(Arg601*) | Paternal | Zhang et al. (2019) [31] |
| Exon 15 | c.2332dupA | p.(Met778Asnfs*4) | Paternal | This report |
| Exon 15 | c.2348C>G | p.(Ser783*) | Homozygous | El-Sayed et al. (2009) [18] |
| Exon 15 | c.2350A>T | p.(Arg784*) | Maternal | Zhang et al. (2019) [31] |
| Exon 15 | c.2522T>A | p.(Leu841Gln) | Maternal | Rungroj et al. (2018) [40] |
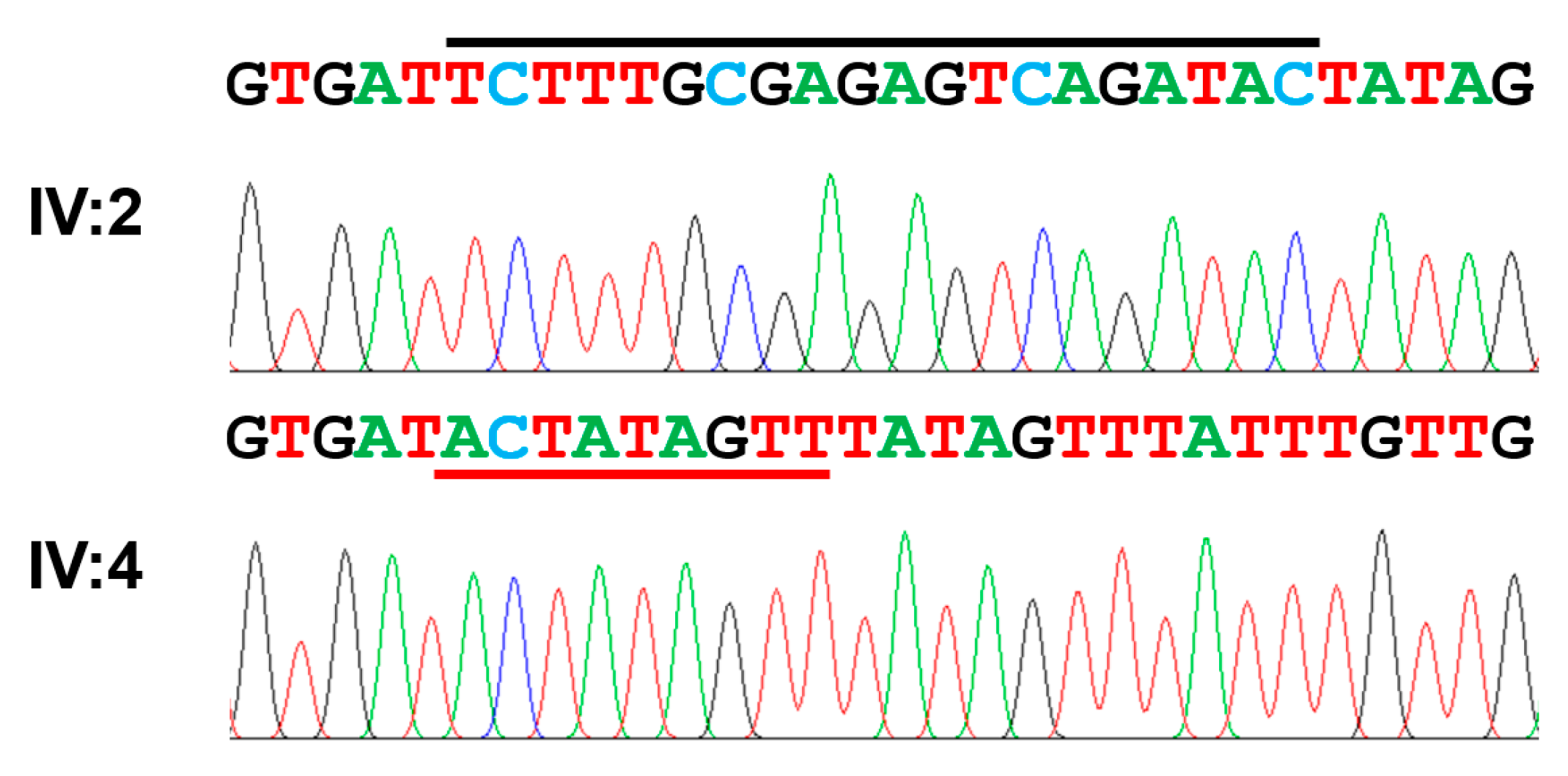
| Exon 15 | c.2680_2699delinsACTATAGTT | p.(Ser894Thrfs*15) | Homozygous | This report |
| Exon 15 | c.2686C>T | p.(Arg896*) | Homozygous | El-Sayed et al. (2011) [45] Rungroj et al. (2018) [40] |
| Exon 16 | c.2857delA | p.(Ser953Valfs*20) | Homozygous | El-Sayed et al. (2009) [18] |
| Exon 16 | c.2866G>A | p.(Arg956*) | Homozygous | Khandelwal et al. (2021) [41] |
| Exon 17 | c.2934G>A | p.(Trp978*) | Homozygous | El-Sayed et al. (2009) [18] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.J.; Zhang, H.; Lee, Y.; Seymen, F.; Koruyucu, M.; Kasimoglu, Y.; Simmer, J.P.; Hu, J.C.-C.; Kim, J.-W. Novel WDR72 Mutations Causing Hypomaturation Amelogenesis Imperfecta. J. Pers. Med. 2023, 13, 326. https://doi.org/10.3390/jpm13020326
Kim YJ, Zhang H, Lee Y, Seymen F, Koruyucu M, Kasimoglu Y, Simmer JP, Hu JC-C, Kim J-W. Novel WDR72 Mutations Causing Hypomaturation Amelogenesis Imperfecta. Journal of Personalized Medicine. 2023; 13(2):326. https://doi.org/10.3390/jpm13020326
Chicago/Turabian StyleKim, Youn Jung, Hong Zhang, Yejin Lee, Figen Seymen, Mine Koruyucu, Yelda Kasimoglu, James P. Simmer, Jan C.-C. Hu, and Jung-Wook Kim. 2023. "Novel WDR72 Mutations Causing Hypomaturation Amelogenesis Imperfecta" Journal of Personalized Medicine 13, no. 2: 326. https://doi.org/10.3390/jpm13020326