
Pharmacogenetics of Cardiovascular Prevention in Diabetes: From Precision Medicine to Identification of Novel Targets


Abstract
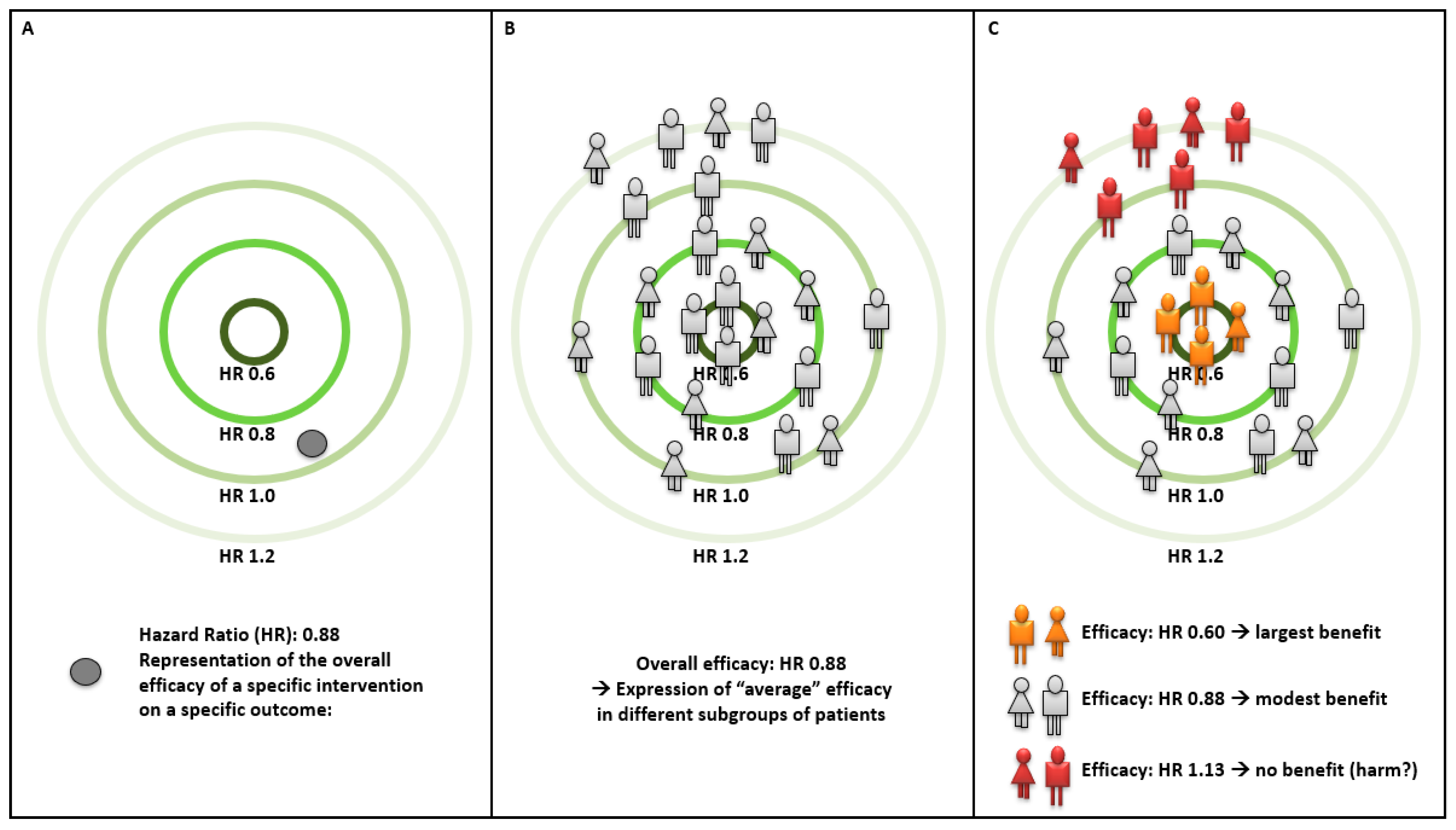
:1. Precision Medicine: Definition and Applications
2. Pharmacogenetic Studies Using Polygenic Risk Score for CAD: CAD-PGRS
2.1. Polygenic Risk Score for CAD to Improve CV Risk Assessments
2.2. Polygenic Risk Score for CAD to Improve Allocation of PCSK9i Treatments
2.3. Polygenic Risk Score for CAD and Allocation of Other Cardiovascular Preventive Treatments
3. Identification of Novel Variants Associated with Different Responses to CV-Preventive Treatments in Diabetes
3.1. Pharmacogenetic Studies to Reduce Adverse Effects of Intensive Glycemic Control
3.2. Pharmacogenetic Studies to Expand The Number of Patients That might Benefit from Treatment: The Example of Pharmacogenetic Studies of Fenofibrate
4. Searching for Diabetes-Specific CAD Genes to Develop Novel Precision Medicine Treatment
5. Advantages and Current Gaps in Using Genetic Variants as Biomarkers for Precision Medicine
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Literature Review Section
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2021, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Rawshani, A.; Rawshani, A.; Franzén, S.; Eliasson, B.; Svensson, A.-M.; Miftaraj, M.; McGuire, D.K.; Sattar, N.; Rosengren, A.; Gudbjörnsdottir, S. Mortality and Cardiovascular Disease in Type 1 and Type 2 Diabetes. N. Engl. J. Med. 2017, 376, 1407–1418. [Google Scholar] [CrossRef]
- Pearson-Stuttard, J.; Bennett, J.; Cheng, Y.J.; Vamos, E.P.; Cross, A.J.; Ezzati, M.; Gregg, E.W. Trends in predominant causes of death in individuals with and without diabetes in England from 2001 to 2018: An epidemiological analysis of linked primary care records. Lancet Diabetes Endocrinol. 2021, 9, 165–173. [Google Scholar] [CrossRef]
- Heald, A.H.; Stedman, M.; Davies, M.; Livingston, M.; Alshames, R.; Lunt, M.; Rayman, G.; Gadsby, R. Estimating life years lost to diabetes: Outcomes from analysis of National Diabetes Audit and Office of National Statistics data. Cardiovasc. Endocrinol. Metab. 2020, 9, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.K.; Suarez-Ortegon, M.F.; Read, S.H.; Kontopantelis, E.; Buchan, I.; Emsley, R.; Sattar, N.; Ashcroft, D.M.; Wild, S.H.; Rutter, M.K. Risk Factor Control and Cardiovascular Event Risk in People With Type 2 Diabetes in Primary and Secondary Prevention Settings. Circulation 2020, 142, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Sattar, N.; Lee, M.M.Y.; Kristensen, S.L.; Branch, K.R.H.; Del Prato, S.; Khurmi, N.S.; Lam, C.S.P.; Lopes, R.D.; McMurray, J.J.V.; Pratley, R.E.; et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of randomised trials. Lancet Diabetes Endocrinol. 2021, 9, 653–662. [Google Scholar] [CrossRef]
- McGuire, D.K.; Shih, W.J.; Cosentino, F.; Charbonnel, B.; Cherney, D.Z.I.; Dagogo-Jack, S.; Pratley, R.; Greenberg, M.; Wang, S.; Huyck, S.; et al. Association of SGLT2 Inhibitors With Cardiovascular and Kidney Outcomes in Patients With Type 2 Diabetes: A Meta-analysis. JAMA Cardiol. 2021, 6, 148–158. [Google Scholar] [CrossRef]
- Chung, W.K.; Erion, K.; Florez, J.C.; Hattersley, A.T.; Hivert, M.-F.; Lee, C.G.; McCarthy, M.I.; Nolan, J.J.; Norris, J.M.; Pearson, E.R.; et al. Precision Medicine in Diabetes: A Consensus Report From the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2020, 43, 1617–1635. [Google Scholar] [CrossRef]
- Hartiala, J.; Schwartzman, W.S.; Gabbay, J.; Ghazalpour, A.; Bennett, B.J.; Allayee, H. The Genetic Architecture of Coronary Artery Disease: Current Knowledge and Future Opportunities. Curr. Atheroscler. Rep. 2017, 19, 6. [Google Scholar] [CrossRef] [Green Version]
- Bodmer, W.; Bonilla, C. Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 2008, 40, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Kathiresan, S. Genetics of Common, Complex Coronary Artery Disease. Cell 2019, 177, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Parast, L.; Cai, T.; Powers, C.; Gervino, E.V.; Hauser, T.H.; Hu, F.B.; Doria, A. Genetic susceptibility to coronary heart disease in type 2 diabetes: 3 independent studies. J. Am. Coll. Cardiol. 2011, 58, 2675–2682. [Google Scholar] [CrossRef] [PubMed]
- Look ARG. Prospective association of a genetic risk score and lifestyle intervention with cardiovascular morbidity and mortality among individuals with type 2 diabetes: The Look AHEAD randomised controlled trial. Diabetologia 2015, 58, 1803–1813. [Google Scholar] [CrossRef]
- Raffield, L.M.; Cox, A.J.; Carr, J.J.; Freedman, B.I.; Hicks, P.J.; Langefeld, C.D.; Hsu, F.-C.; Bowden, D.W. Analysis of a cardiovascular disease genetic risk score in the Diabetes Heart Study. Acta Diabetol. 2015, 52, 743–751. [Google Scholar] [CrossRef]
- Morieri, M.L.; Gao, H.; Pigeyre, M.; Shah, H.S.; Sjaarda, J.; Mendonca, C.; Hastings, T.; Buranasupkajorn, P.; Motsinger-Reif, A.A.; Rotroff, D.M.; et al. Genetic Tools for Coronary Risk Assessment in Type 2 Diabetes: A Cohort Study From the ACCORD Clinical Trial. Diabetes Care 2018, 41, 2404–2413. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, J.; Haloui, M.; Attaoua, R.; Tahir, R.; Hishmih, C.; Harvey, F.; Marois-Blanchet, F.-C.; Long, C.; Simon, P.; Santucci, L.; et al. Polygenic risk scores predict diabetes complications and their response to intensive blood pressure and glucose control. Diabetologia 2021, 64, 2012–2025. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.G.; Rader, D.J. Polygenic Risk Scores and Coronary Artery Disease: Ready for Prime Time? Circulation 2020, 141, 637–640. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Zekavat, S.; Collins, R.L.; Roselli, C.; Natarajan, P.; Lichtman, J.H.; D’Onofrio, G.; Mattera, J.A.; Dreyer, R.P.; et al. Whole-Genome Sequencing to Characterize Monogenic and Polygenic Contributions in Patients Hospitalized with Early-Onset Myocardial Infarction. Circulation 2019, 139, 1593–1602. [Google Scholar] [CrossRef] [Green Version]
- Torkamani, A.; Wineinger, N.E.; Topol, E.J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 2018, 19, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Marston, N.A.; Kamanu, F.; Nordio, F.; Gurmu, Y.; Roselli, C.; Sever, P.S.; Pedersen, T.R.; Keech, A.C.; Wang, H.; Pineda, A.L.; et al. Predicting Benefit From Evolocumab Therapy in Patients With Atherosclerotic Disease Using a Genetic Risk Score: Results From the FOURIER Trial. Circulation 2020, 141, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Damask, A.; Steg, P.G.; Schwartz, G.G.; Szarek, M.; Hagström, E.; Badimon, L.; Chapman, M.J.; Boileau, C.; Tsimikas, S.; Ginsberg, H.N.; et al. Patients With High Genome-Wide Polygenic Risk Scores for Coronary Artery Disease May Receive Greater Clinical Benefit From Alirocumab Treatment in the ODYSSEY OUTCOMES Trial. Circulation 2020, 141, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Brégeault, M.-F.; Dalby, A.J.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; et al. Effect of Alirocumab on Mortality After Acute Coronary Syndromes. Circulation 2019, 140, 103–112. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Arca, M.; Ansell, D.; Averna, M.; Fanelli, F.; Gorcyca, K.; Iorga, R.; Maggioni, A.P.; Paizis, G.; Tomic, R.; Catapano, A.L. Statin utilization and lipid goal attainment in high or very-high cardiovascular risk patients: Insights from Italian general practice. Atherosclerosis 2018, 271, 120–127. [Google Scholar] [CrossRef]
- Morieri, M.L.; Avogaro, A.; Fadini, G.P. Cholesterol lowering therapies and achievement of targets for primary and secondary cardiovascular prevention in type 2 diabetes: Unmet needs in a large population of outpatients at specialist clinics. Cardiovasc. Diabetol. 2020, 19, 190. [Google Scholar] [CrossRef]
- Ray, K.K.; Molemans, B.; Schoonen, W.M.; Giovas, P.; Bray, S.; Kiru, G.; Murphy, J.; Banach, M.; De Servi, S.; Gaita, D.; et al. EU-Wide Cross-Sectional Observational Study of Lipid-Modifying Therapy Use in Secondary and Primary Care: The DA VINCI study. Eur. J. Prev. Cardiol. 2020, 28, 1279–1289. [Google Scholar] [CrossRef]
- Abushanab, D.; Al-Badriyeh, D.; Marquina, C.; Bailey, C.; Jaam, M.; Liew, D.; Ademi, Z. A Systematic Review of Cost-Effectiveness of Non-Statin Lipid-Lowering Drugs for Primary and Secondary Prevention of Cardiovascular Disease in Patients with Type 2 Diabetes Mellitus. Curr. Probl. Cardiol. 2022, 101211. [Google Scholar] [CrossRef]
- Natarajan, P.; Young, R.; Stitziel, N.O.; Padmanabhan, S.; Baber, U.; Mehran, R.; Sartori, S.; Fuster, V.; Reilly, D.F.; Butterworth, A.; et al. Polygenic Risk Score Identifies Subgroup With Higher Burden of Atherosclerosis and Greater Relative Benefit From Statin Therapy in the Primary Prevention Setting. Circulation 2017, 135, 2091–2101. [Google Scholar] [CrossRef] [Green Version]
- Mega, J.L.; Stitziel, N.O.; Smith, J.G.; Chasman, D.I.; Caulfield, M.; Devlin, J.J.; Nordio, F.; Hyde, C.L.; Cannon, C.P.; Sacks, F.M.; et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: An analysis of primary and secondary prevention trials. Lancet 2015, 385, 2264–2271. [Google Scholar] [CrossRef]
- Tikkanen, E.; Havulinna, A.S.; Palotie, A.; Salomaa, V.; Ripatti, S. Genetic risk prediction and a 2-stage risk screening strategy for coronary heart disease. Arter. Thromb. Vasc. Biol. 2013, 33, 2261–2266. [Google Scholar] [CrossRef] [PubMed]
- Kullo, I.J.; Jouni, H.; Austin, E.E.; Brown, S.-A.; Kruisselbrink, T.M.; Isseh, I.N.; Haddad, R.A.; Marroush, T.S.; Shameer, K.; Olson, J.E.; et al. Incorporating a Genetic Risk Score Into Coronary Heart Disease Risk Estimates: Effect on Low-Density Lipoprotein Cholesterol Levels (the MI-GENES Clinical Trial). Circulation 2016, 133, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.D.; Schonman, E.F.; Robinson, J.O.; Roberts, J.S.; Diamond, P.M.; Lee, K.B.; Green, R.C.; McGuire, A.L. Behavioral and psychological impact of genome sequencing: A pilot randomized trial of primary care and cardiology patients. NPJ Genom. Med. 2021, 6, 72. [Google Scholar] [CrossRef]
- Morieri, M.L.; Shah, H.; Doria, A.; the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Genetic Study Group. Variants in ANGPTL4 and the Risk of Coronary Artery Disease. N. Engl. J. Med. 2016, 375, 2304–2305. [Google Scholar] [CrossRef]
- Duval, C.; Muller, M.; Kersten, S. PPARalpha and dyslipidemia. Biochim. Et Biophys. Acta 2007, 1771, 961–971. [Google Scholar] [CrossRef]
- Morieri, M.L.; Shah, H.S.; Sjaarda, J.; Lenzini, P.A.; Campbell, H.; Motsinger-Reif, A.A.; Gao, H.; Lovato, L.; Prudente, S.; Pandolfi, A.; et al. PPARA Polymorphism Influences the Cardiovascular Benefit of Fenofibrate in Type 2 Diabetes: Findings From ACCORD-Lipid. Diabetes 2020, 69, 771–783. [Google Scholar] [CrossRef]
- Shah, H.S.; Gao, H.; Morieri, M.L.; Skupien, J.; Marvel, S.; Paré, G.; Mannino, G.C.; Buranasupkajorn, P.; Mendonca, C.; Hastings, T.; et al. Genetic Predictors of Cardiovascular Mortality During Intensive Glycemic Control in Type 2 Diabetes: Findings From the ACCORD Clinical Trial. Diabetes Care 2016, 39, 1915–1924. [Google Scholar] [CrossRef]
- Shah, H.S.; Morieri, M.L.; Marcovina, S.M.; Sigal, R.J.; Gerstein, H.C.; Wagner, M.J.; Motsinger-Reif, A.A.; Buse, J.B.; Kraft, P.; Mychaleckyj, J.C.; et al. Modulation of GLP-1 Levels by a Genetic Variant That Regulates the Cardiovascular Effects of Intensive Glycemic Control in ACCORD. Diabetes Care 2018, 41, 348–355. [Google Scholar] [CrossRef]
- Ray, K.K.; Seshasai, S.R.K.; Wijesuriya, S.; Sivakumaran, R.; Nethercott, S.; Preiss, D.; Erqou, S.; Sattar, N. Effect of intensive control of glucose on cardiovascular outcomes and death in patients with diabetes mellitus: A meta-analysis of randomised controlled trials. Lancet 2009, 373, 1765–1772. [Google Scholar] [CrossRef]
- Skyler, J.S.; Bergenstal, R.; Bonow, R.O.; Buse, J.; Deedwania, P.; Gale, E.A.; Howard, B.V.; Kirkman, M.S.; Kosiborod, M.; Reaven, P.; et al. Intensive glycemic control and the prevention of cardiovascular events: Implications of the ACCORD, ADVANCE, and VA diabetes trials: A position statement of the American Diabetes Association and a scientific statement of the American College of Cardiology Foundation and the American Heart Association. Circulation 2009, 119, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein, H.C.; Miller, M.E.; Byington, R.P.; Goff, D.C., Jr.; Bigger, J.T.; Buse, J.B.; Cushman, W.C.; Genuth, S.; Ismail-Beigi, F.; et al. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 2008, 358, 2545–2559. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Aguiar, C.; Alegria, E.; Bonadonna, R.C.; Cosentino, F.; Elisaf, M.; Farnier, M.; Ferrieres, J.; Filardi, P.P.; Hancu, N.; et al. Current practice in identifying and treating cardiovascular risk, with a focus on residual risk associated with atherogenic dyslipidaemia. Eur. Heart J. Suppl. 2016, 18 (Suppl. C), C2–C12. [Google Scholar] [CrossRef]
- Taskinen, M.R.; Boren, J. New insights into the pathophysiology of dyslipidemia in type 2 diabetes. Atherosclerosis 2015, 239, 483–495. [Google Scholar] [CrossRef]
- Ginsberg, H.N.; Packard, C.J.; Chapman, M.J.; Borén, J.; Aguilar-Salinas, C.A.; Averna, M.; Ference, B.A.; Gaudet, D.; Hegele, R.A.; Kersten, S.; et al. Triglyceride-rich lipoproteins and their remnants: Metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies-a consensus statement from the European Atherosclerosis Society. Eur. Heart J. 2021, 42, 4791–4806. [Google Scholar] [CrossRef]
- American Diabetes Association 10. Cardiovascular Disease and Risk Management: Standards of Medical Care in Diabetes-2021. Diabetes Care 2021, 44 (Suppl. S1), S125–S150. [Google Scholar] [CrossRef]
- Jun, M.; Foote, C.; Lv, J.; Neal, B.; Patel, A.; Nicholls, S.J.; Grobbee, D.E.; Cass, A.; Chalmers, J.; Perkovic, V. Effects of fibrates on cardiovascular outcomes: A systematic review and meta-analysis. Lancet 2010, 375, 1875–1884. [Google Scholar] [CrossRef]
- Sacks, F.M.; Carey, V.J.; Fruchart, J.C. Combination lipid therapy in type 2 diabetes. N. Engl. J. Med. 2010, 363, 692–694. [Google Scholar]
- Bruckert, E.; Labreuche, J.; Deplanque, D.; Touboul, P.J.; Amarenco, P. Fibrates effect on cardiovascular risk is greater in patients with high triglyceride levels or atherogenic dyslipidemia profile: A systematic review and meta-analysis. J. Cardiovasc. Pharmacol. 2011, 57, 267–272. [Google Scholar] [CrossRef]
- Kim, N.H.; Han, K.H.; Choi, J.; Lee, J.; Kim, S.G. Use of fenofibrate on cardiovascular outcomes in statin users with metabolic syndrome: Propensity matched cohort study. BMJ 2019, 366, l5125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T., Jr.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Morieri, M.L. Heart Failure Burden in Diabetes: Can Fenofibrate Provide Additional Hope? Diabetes Care 2022, 45, 1500–1502. [Google Scholar] [CrossRef] [PubMed]
- Pipino, C.; Shah, H.; Prudente, S.; Di Pietro, N.; Zeng, L.; Park, K.; Trischitta, V.; Pennathur, S.; Pandolfi, A.; Doria, A. Association of the 1q25 Diabetes-Specific Coronary Heart Disease Locus With Alterations of the gamma-Glutamyl Cycle and Increased Methylglyoxal Levels in Endothelial Cells. Diabetes 2020, 69, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Qi, Q.; Prudente, S.; Mendonca, C.; Andreozzi, F.; Di Pietro, N.; Sturma, M.; Novelli, V.; Mannino, G.C.; Formoso, G.; et al. Association between a genetic variant related to glutamic acid metabolism and coronary heart disease in individuals with type 2 diabetes. JAMA 2013, 310, 821–828. [Google Scholar] [CrossRef]
- Look ARG. Prospective Association of GLUL rs10911021 with Cardiovascular Morbidity and Mortality Among Individuals With Type 2 Diabetes: The Look AHEAD Study. Diabetes 2016, 65, 297–302. [Google Scholar] [CrossRef]
- Prudente, S.; Shah, H.; Bailetti, D.; Pezzolesi, M.; Buranasupkajorn, P.; Mercuri, L.; Mendonca, C.; De Cosmo, S.; Niewczas, M.; Trischitta, V.; et al. Genetic Variant at the GLUL Locus Predicts All-Cause Mortality in Patients with Type 2 Diabetes. Diabetes 2015, 64, 2658–2663. [Google Scholar] [CrossRef]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 226–235. [Google Scholar] [CrossRef]
- Niihara, Y.; Zerez, C.R.; Akiyama, D.S.; Tanaka, K.R. Oral L-glutamine therapy for sickle cell anemia: I. Subjective clinical improvement and favorable change in red cell NAD redox potential. Am. J. Hematol. 1998, 58, 117–121. [Google Scholar] [CrossRef]
- Morrow, D.A.; de Lemos, J.A. Benchmarks for the assessment of novel cardiovascular biomarkers. Circulation 2007, 115, 949–952. [Google Scholar] [CrossRef]
- Verbelen, M.; Weale, M.E.; Lewis, C.M. Cost-effectiveness of pharmacogenetic-guided treatment: Are we there yet? Pharm. J. 2017, 17, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vujkovic, M.; Keaton, J.M.; Lynch, J.A.; Miller, D.R.; Zhou, J.; Tcheandjieu, C.; Huffman, J.E.; Assimes, T.L.; Lorenz, K.; Zhu, X.; et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 2020, 52, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Gurdasani, D.; Barroso, I.; Zeggini, E.; Sandhu, M.S. Genomics of disease risk in globally diverse populations. Nat. Rev. Genet. 2019, 20, 520–535. [Google Scholar] [CrossRef]
- Pereira, L.; Mutesa, L.; Tindana, P.; Ramsay, M. African genetic diversity and adaptation inform a precision medicine agenda. Nat. Rev. Genet. 2021, 22, 284–306. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP | Allele 1/2 | White | African American | Meta-Analysis p Value | rs6008845 Conditional Analysis p Value | ||||
|---|---|---|---|---|---|---|---|---|---|
| p Value | Effect ± S.E. | Allele1 Freq | p Value | Effect ± S.E. | Allele1 Freq | ||||
| rs6008845 | c/t | 3.7 × 10−4 | 0.59 ± 0.17 | 0.40 | 2.4 × 10−2 | 1.39 ± 0.62 | 0.79 | 5.6 × 10−5 | ref |
| rs135557 | g/a | 9.3 × 10−4 | 0.54 ± 0.16 | 0.44 | 6.8 × 10−3 | 1.43 ± 0.53 | 0.72 | 7.3 × 10−5 | 3.0 × 10−1 |
| rs135570 | g/a | 9.0 × 10−4 | 0.54 ± 0.16 | 0.46 | 5.3 × 10−3 | 2.16 ± 0.77 | 0.83 | 1.3 × 10−4 | 5.4 × 10−1 |
| rs6007904 | g/a | 6.4 × 10−4 | 0.56 ± 0.16 | 0.42 | 1.9 × 10−1 | 0.71 ± 0.54 | 0.73 | 2.6 × 10−4 | 9.4 × 10−1 |
| rs2105914 | g/a | 1.9 × 10−3 | 0.50 ± 0.16 | 0.48 | 2.7 × 10−2 | 1.74 ± 0.79 | 0.83 | 5.0 × 10−4 | 6.8 × 10−1 |
| rs135577 | a/g | 2.2 × 10−3 | 0.56 ± 0.18 | 0.28 | 1.1 × 10−1 | 0.70 ± 0.44 | 0.57 | 5.8 × 10−4 | 6.4 × 10−1 |
| rs11090910 | c/t | 8.1 × 10−3 | 0.49 ± 0.18 | 0.27 | 3.5 × 10−2 | 0.89 ± 0.42 | 0.41 | 1.1 × 10−3 | 4.2 × 10−2 |
| rs9615904 | c/t | 1.2 × 10−2 | 0.46 ± 0.18 | 0.29 | 2.9 × 10−2 | 1.15 ± 0.52 | 0.81 | 2.1 × 10−3 | 1.3 × 10−2 |
| rs4508712 | g/a | 1.6 × 10−2 | 0.43 ± 0.18 | 0.29 | 2.1 × 10−2 | 1.18 ± 0.51 | 0.81 | 2.4 × 10−3 | 1.2 × 10−2 |
| rs9615264 | a/g | 2.6 × 10−3 | 0.95 ± 0.32 | 0.09 | 9.6 × 10−1 | 0.08 ± 1.59 | 0.02 | 3.1 × 10−3 | 2.3 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morieri, M.L.; Pipino, C.; Doria, A. Pharmacogenetics of Cardiovascular Prevention in Diabetes: From Precision Medicine to Identification of Novel Targets. J. Pers. Med. 2022, 12, 1402. https://doi.org/10.3390/jpm12091402
Morieri ML, Pipino C, Doria A. Pharmacogenetics of Cardiovascular Prevention in Diabetes: From Precision Medicine to Identification of Novel Targets. Journal of Personalized Medicine. 2022; 12(9):1402. https://doi.org/10.3390/jpm12091402
Chicago/Turabian StyleMorieri, Mario Luca, Caterina Pipino, and Alessandro Doria. 2022. "Pharmacogenetics of Cardiovascular Prevention in Diabetes: From Precision Medicine to Identification of Novel Targets" Journal of Personalized Medicine 12, no. 9: 1402. https://doi.org/10.3390/jpm12091402