
Reducing the Excess Activin Signaling Rescues Muscle Degeneration in Myotonic Dystrophy Type 2 Drosophila Model

Abstract
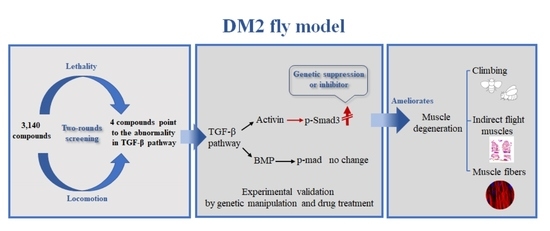
:
1. Introduction
2. Materials and Methods
2.1. Drosophila Stocks
2.2. Drug Treatment
2.3. Climbing Assays
2.4. Lifespan Analysis
2.5. Immunolabeling and Confocal Microscopy
2.6. Drosophila Paraffin Section
2.7. Western Blot
2.8. Statistical Analysis
3. Results
3.1. Drugs Targeting the TGF-β Pathway Could Rescue Lethality and Locomotion of DM2 Fly Model
3.2. Flies Expressing CCTG Repeats in the Muscle at the Adult Stage Showed Shorter Lifespan and Muscle Degeneration
3.3. The Activin Signaling Is Increased in the DM2 Fly Model
3.4. Reducing Activin Signaling Could Rescue Muscle Degeneration
3.5. Reducing Activin Signaling Could Rescue Myofiber Defects
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buxton, J.; Shelbourne, P.; Davies, J.; Jones, C.; Van Tongeren, T.; Aslanidis, C.; de Jong, P.; Jansen, G.; Anvret, M.; Riley, B.; et al. Detection of an unstable fragment of DNA specific to individuals with myotonic dystrophy. Nature 1992, 355, 547–548. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barceló, J.; O’Hoy, K.; et al. Myotonic dystrophy mutation: An unstable CTG repeat in the 3′ untranslated region of the gene. Science 1992, 255, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.H.; Pizzuti, A.; Fenwick, R.G.; King, J.; Rajnarayan, S.; Dunne, P.W.; Dubel, J.; Nasser, G.A.; Ashizawa, T.; de Jong, P.; et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992, 255, 1256–1258. [Google Scholar] [CrossRef] [PubMed]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [Green Version]
- Botta, A.; Caldarola, S.; Vallo, L.; Bonifazi, E.; Fruci, D.; Gullotta, F.; Massa, R.; Novelli, G.; Loreni, F. Effect of the [CCTG]n repeat expansion on ZNF9 expression in myotonic dystrophy type II (DM2). Biochim. Biophys. Acta 2006, 1762, 329–334. [Google Scholar] [CrossRef]
- Kalsotra, A.; Xiao, X.; Ward, A.J.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. USA 2008, 105, 20333–20338. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.P.; Nakamori, M.; Mochizuki, H. Therapeutic development in myotonic dystrophy. Rinsho Shinkeigaku 2014, 54, 1077–1079. [Google Scholar] [CrossRef] [Green Version]
- Meola, G.; Cardani, R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Biophys. Acta 2015, 1852, 594–606. [Google Scholar] [CrossRef] [Green Version]
- Raheem, O.; Olufemi, S.E.; Bachinski, L.L.; Vihola, A.; Sirito, M.; Holmlund-Hampf, J.; Haapasalo, H.; Li, Y.P.; Udd, B.; Krahe, R. Mutant (CCTG)n expansion causes abnormal expression of zinc finger protein 9 (ZNF9) in myotonic dystrophy type 2. Am. J. Pathol. 2010, 177, 3025–3036. [Google Scholar] [CrossRef]
- Timchenko, N.A.; Cai, Z.-J.; Welm, A.L.; Reddy, S.; Ashizawa, T.; Timchenko, L.T. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J. Biol. Chem. 2001, 276, 7820–7826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, A.J.; Rimer, M.; Killian, J.M.; Dowling, J.J.; Cooper, T.A. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum. Mol. Genet. 2010, 19, 3614–3622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, K.; Jin, B.; Iakova, P.; Huichalaf, C.; Sarkar, P.; Schneider-Gold, C.; Schoser, B.; Meola, G.; Shyu, A.-B.; Timchenko, N.; et al. RNA Foci, CUGBP1, and ZNF9 are the primary targets of the mutant CUG and CCUG repeats expanded in myotonic dystrophies type 1 and type 2. Am. J. Pathol. 2011, 179, 2475–2489. [Google Scholar] [CrossRef] [PubMed]
- Sellier, C.; Cerro-Herreros, E.; Blatter, M.; Freyermuth, F.; Gaucherot, A.; Ruffenach, F.; Sarkar, P.; Puymirat, J.; Udd, B.; Day, J.W.; et al. rbFOX1/MBNL1 competition for CCUG RNA repeats binding contributes to myotonic dystrophy type 1/type 2 differences. Nat. Commun. 2018, 9, 2009. [Google Scholar] [CrossRef] [PubMed]
- Mooers, B.H.; Logue, J.S.; Berglund, J.A. The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proc. Natl. Acad. Sci. USA 2005, 102, 16626–16631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Matsumoto, J.; Bai, L.-P.; Murata, A.; Dohno, C.; Nakatani, K. A Ligand That Targets CUG Trinucleotide Repeats. Chemistry 2016, 22, 14881–14889. [Google Scholar] [CrossRef]
- Li, J.; Nakamori, M.; Matsumoto, J.; Murata, A.; Dohno, C.; Kiliszek, A.; Taylor, K.; Sobczak, K.; Nakatani, K. A Dimeric 2,9-Diamino-1,10-phenanthroline Derivative Improves Alternative Splicing in Myotonic Dystrophy Type 1 Cell and Mouse Models. Chemistry 2018, 24, 18115–18122. [Google Scholar] [CrossRef]
- Zhang, F.; Bodycombe, N.E.; Haskell, K.M.; Sun, Y.L.; Wang, E.T.; Morris, C.A.; Jones, L.H.; Wood, L.D.; Pletcher, M.T. A flow cytometry-based screen identifies MBNL1 modulators that rescue splicing defects in myotonic dystrophy type I. Hum. Mol. Genet. 2017, 26, 3056–3068. [Google Scholar] [CrossRef] [Green Version]
- Oana, K.; Oma, Y.; Suo, S.; Takahashi, M.P.; Nishino, I.; Takeda, S.; Ishiura, S. Manumycin A corrects aberrant splicing of Clcn1 in myotonic dystrophy type 1 (DM1) mice. Sci. Rep. 2013, 3, 2142. [Google Scholar] [CrossRef]
- Wang, G.-S.; Kuyumcu-Martinez, M.N.; Sarma, S.; Mathur, N.; Wehrens, X.; Cooper, T.A. PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J. Clin. Investig. 2009, 119, 3797–3806. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, M.; Selma-Soriano, E.; Magny, E.; Couso, J.P.; Pérez-Alonso, M.; Charlet, N.; Artero, R.; Llamusi, B. Pentamidine rescues contractility and rhythmicity in a Drosophila model of myotonic dystrophy heart dysfunction. Dis. Models Mech. 2015, 8, 1569–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yenigun, V.B.; Sirito, M.; Amcheslavky, A.; Czernuszewicz, T.; Colonques-Bellmunt, J.; García-Alcover, I.; Wojciechowska, M.; Bolduc, C.; Chen, Z.; Castel, A.L.; et al. (CCUG)n RNA toxicity in a Drosophila model of myotonic dystrophy type 2 (DM2) activates apoptosis. Dis. Models Mech. 2017, 10, 993–1003. [Google Scholar] [CrossRef] [Green Version]
- Qurashi, A.; Liu, H.; Ray, L.; Nelson, D.L.; Duan, R.; Jin, P. Chemical screen reveals small molecules suppressing fragile X premutation rCGG repeat-mediated neurodegeneration in Drosophila. Hum. Mol. Genet. 2012, 21, 2068–2075. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Hagemann, T.L.; Messing, A.; Feany, M.B. An In Vivo Pharmacological Screen Identifies Cholinergic Signaling as a Therapeutic Target in Glial-Based Nervous System Disease. J. Neurosci. 2016, 36, 1445–1455. [Google Scholar] [CrossRef] [Green Version]
- Palandri, A.; Martin, E.; Russi, M.; Rera, M.; Tricoire, H.; Monnier, V. Identification of cardioprotective drugs by medium-scale in vivo pharmacological screening on a Drosophila cardiac model of Friedreich’s ataxia. Dis. Models Mech. 2018, 11, dmm033811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Goodman, L.; Shieh, S.-Y.; Min, M.; Teng, X.; Zhu, Y.; Bonini, N.M. A fly model for the CCUG-repeat expansion of myotonic dystrophy type 2 reveals a novel interaction with MBNL1. Hum. Mol. Genet. 2015, 24, 954–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, A.; Moss-Taylor, L.; Kim, M.J.; Ghosh, A.C.; O’Connor, M.B. TGF-beta Family Signaling in Drosophila. Cold Spring Harb. Perspect. Biol. 2017, 9, a022152. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.L.; Colgan, T.D.; Walton, K.L.; Gregorevic, P.; Harrison, C.A. The TGF-beta Signalling Network in Muscle Development, Adaptation and Disease. Adv. Exp. Med. Biol. 2016, 900, 97–131. [Google Scholar]
- Caygill, E.E.; Brand, A.H. The GAL4 System: A Versatile System for the Manipulation and Analysis of Gene Expression. Methods Mol. Biol. 2016, 1478, 33–52. [Google Scholar]
- Kucherenko, M.M.; Marrone, A.K.; Rishko, V.M.; Yatsenko, A.S.; Klepzig, A.; Shcherbata, H.R. Paraffin-embedded and frozen sections of Drosophila adult muscles. J. Vis. Exp. 2010, 46, 2438. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.R.; Liu, X.; Lee, L.; Laron, D.; Ning, A.Y.; Kim, H.T.; Feeley, B.T. TGF-beta Small Molecule Inhibitor SB431542 Reduces Rotator Cuff Muscle Fibrosis and Fatty Infiltration by Promoting Fibro/Adipogenic Progenitor Apoptosis. PLoS ONE 2016, 11, e0155486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, K.; Yong, C.W.; Hua, K. SB431542 inhibited cigarette smoke extract induced invasiveness of A549 cells via the TGF-beta1/Smad2/MMP3 pathway. Oncol. Lett. 2018, 15, 9681–9686. [Google Scholar] [PubMed] [Green Version]
- Shou, J.; Cao, J.; Zhang, S.; Sun, R.; Zhao, M.; Chen, K.; Su, S.B.; Yang, J.; Yang, T. SIS3, a specific inhibitor of smad3, attenuates bleomycin-induced pulmonary fibrosis in mice. Biochem. Biophys. Res. Commun. 2018, 503, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Hermans, M.; Pinto, Y.; Merkies, I.; de Die-Smulders, C.; Crijns, H.; Faber, C.G. Hereditary muscular dystrophies and the heart. Neuromuscul. Disord. 2010, 20, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Delaney, K.; Kasprzycka, P.; Ciemerych, M.A.; Zimowska, M. The role of TGF-beta1 during skeletal muscle regeneration. Cell Biol. Int. 2017, 41, 706–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaden, B.C.; Wang, Y.X.; Wilson, J.M.; Culver, A.E.; Milner, A.; Datta-Mannan, A.; Shetler, P.; Croy, J.E.; Dai, G.; Krishnan, V. Inhibition of activin A ameliorates skeletal muscle injury and rescues contractile properties by inducing efficient remodeling in female mice. Am. J. Pathol. 2014, 184, 1152–1166. [Google Scholar] [CrossRef]
- Stewart, J.D.; Masi, T.L.; Cumming, A.E.; Molnar, G.M.; Wentworth, B.M.; Sampath, K.; McPherson, J.M.; Yaeger, P.C. Characterization of proliferating human skeletal muscle-derived cells in vitro: Differential modulation of myoblast markers by TGF-beta2. J. Cell Physiol. 2003, 196, 70–78. [Google Scholar] [CrossRef]
- Rathbone, C.R.; Yamanouchi, K.; Chen, X.K.; Nevoret-Bell, C.J.; Rhoads, R.P.; Allen, R.E. Effects of transforming growth factor-beta (TGF-beta1) on satellite cell activation and survival during oxidative stress. J. Muscle Res. Cell Motil. 2011, 32, 99–109. [Google Scholar] [CrossRef]
- Lichtman, M.K.; Otero-Vinas, M.; Falanga, V. Transforming growth factor beta (TGF-beta) isoforms in wound healing and fibrosis. Wound Repair Regen. 2016, 24, 215–222. [Google Scholar] [CrossRef]
- Wallace, G.Q.; McNally, E.M. Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu. Rev. Physiol. 2009, 71, 37–57. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J. Mol. Cell Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turillazzi, E.; Neri, M.; Riezzo, I.; Di Paolo, M.; Evangelisti, L.; Fineschi, V. Cardiac fibrosis, arrhythmia and sudden death in myotonic dystrophy type 1: Could TGF-ss1 improve the predictive accuracy of patients at risk, opening new therapeutic challenges? Int. J. Cardiol. 2013, 168, 4976–4978. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yao, S.; Liu, Y.; Long, L.; Yang, H.; Li, Q.; Liang, J.; Li, X.; Lu, Y.; Zhu, H.; et al. Expression levels of TGF-beta1 and CTGF are associated with the severity of Duchenne muscular dystrophy. Exp. Ther. Med. 2017, 13, 1209–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, R.D.; van Erp, C.; Habashi, J.P.; Soleimani, A.A.; Klein, E.C.; Lisi, M.T.; Gamradt, M.; ap Rhys, C.M.; Holm, T.M.; Loeys, B.L.; et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 2007, 13, 204–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, J.A.; Bogdanovich, S.; Beiriger, A.; Wren, L.M.; Rossi, A.E.; Gao, Q.Q.; Gardner, B.; Earley, J.U.; Molkentin, J.D.; McNally, E.M. Excess SMAD signaling contributes to heart and muscle dysfunction in muscular dystrophy. Hum. Mol. Genet. 2014, 23, 6722–6731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasteuning-Vuhman, S.; der Meulen, J.W.B.; Van Putten, M.; Overzier, M.; Dijke, P.T.; Kielbasa, S.M.; Arindrarto, W.; Wolterbeek, R.; Lezhnina, K.V.; Ozerov, I.V.; et al. New function of the myostatin/activin type I receptor (ALK4) as a mediator of muscle atrophy and muscle regeneration. FASEB J. 2017, 31, 238–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, J.A.; Kelly, S.M.; LoPresti, P.P.; Heydemann, A.; Earley, J.U.; Ferguson, E.L.; Wolf, M.J.; McNally, E.M. SMAD signaling drives heart and muscle dysfunction in a Drosophila model of muscular dystrophy. Hum. Mol. Genet. 2011, 20, 894–904. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Sahenk, Z.; Malik, V.; Gomez, A.M.; Flanigan, K.M.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Meadows, E.; Lewis, S.; et al. A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol. Ther. 2015, 23, 192–201. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | PubChem CID | Molecular Formula | Chemical Name |
|---|---|---|---|
| 3952-H8 | 5382764 | C10H8N2O2 | 2-[(E)-2-nitroethenyl]-1H-indole |
| 3972-D11 | 239794 | C21H14ClN3O4S | 4-[(3-chloro-1,4-dioxonaphthalen-2-yl)amino]-N-pyridin-2-ylbenzenesulfonamide |
| 3921-D8 | 364428 | C9H6OS2 | 5-thiophen-2-ylthiophene-2-carbaldehyde |
| 2910-G5 | 319846 | C37H47NO14 | 23-(dimethylamino)-4,8,12,22,24-pentahydroxy-1,12-dimethyl-10-(3,4,5-trimethoxy-4,6-dimethyloxan-2-yl) oxy-20,25-dioxahexacyclo [19.3.1.02,19.05,18.07,16.09,14] pentacosa-2,4,7(16),8,14,18-hexaene-6,17-dione |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, J.; Guan, X.-X.; Zhu, Y.-B.; Deng, H.-T.; Li, G.-X.; Guo, Y.-C.; Jin, P.; Duan, R.-H.; Huang, W. Reducing the Excess Activin Signaling Rescues Muscle Degeneration in Myotonic Dystrophy Type 2 Drosophila Model. J. Pers. Med. 2022, 12, 385. https://doi.org/10.3390/jpm12030385
Deng J, Guan X-X, Zhu Y-B, Deng H-T, Li G-X, Guo Y-C, Jin P, Duan R-H, Huang W. Reducing the Excess Activin Signaling Rescues Muscle Degeneration in Myotonic Dystrophy Type 2 Drosophila Model. Journal of Personalized Medicine. 2022; 12(3):385. https://doi.org/10.3390/jpm12030385
Chicago/Turabian StyleDeng, Jing, Xin-Xin Guan, Ying-Bao Zhu, Hai-Tao Deng, Guang-Xu Li, Yi-Chen Guo, Peng Jin, Ran-Hui Duan, and Wen Huang. 2022. "Reducing the Excess Activin Signaling Rescues Muscle Degeneration in Myotonic Dystrophy Type 2 Drosophila Model" Journal of Personalized Medicine 12, no. 3: 385. https://doi.org/10.3390/jpm12030385