
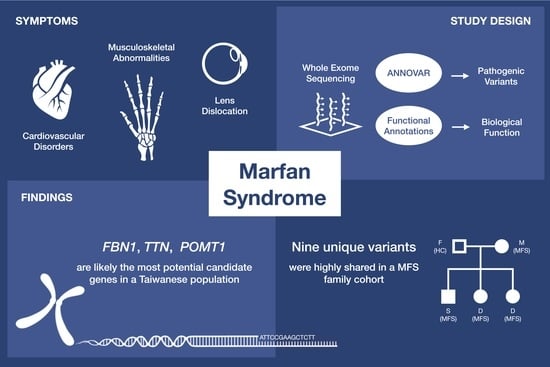
Application of Whole Exome Sequencing and Functional Annotations to Identify Genetic Variants Associated with Marfan Syndrome

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Results
2.1. Patient Basal Characteristic
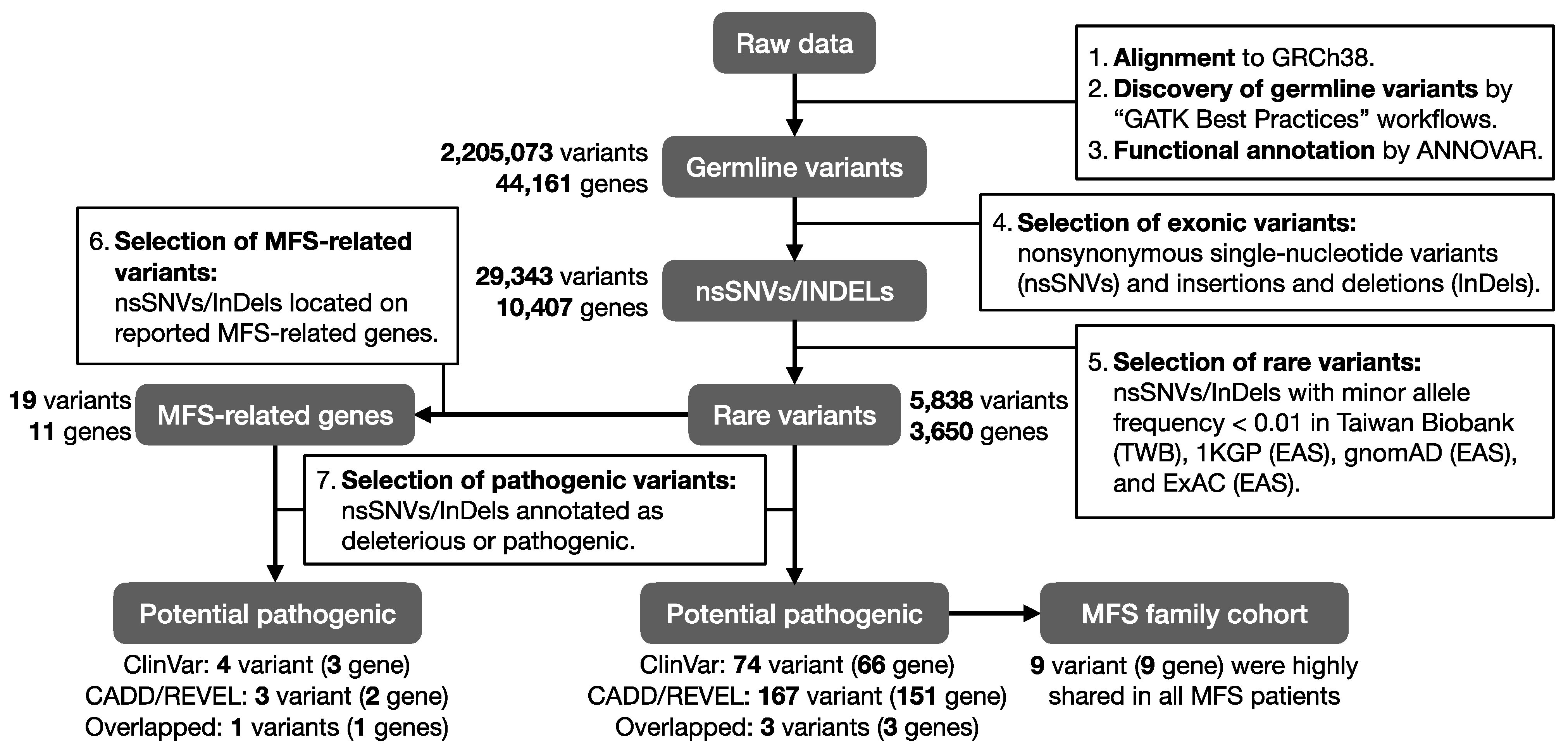
2.2. Pathogenic Rare Variants on Previously Reported MFS-Related Genes
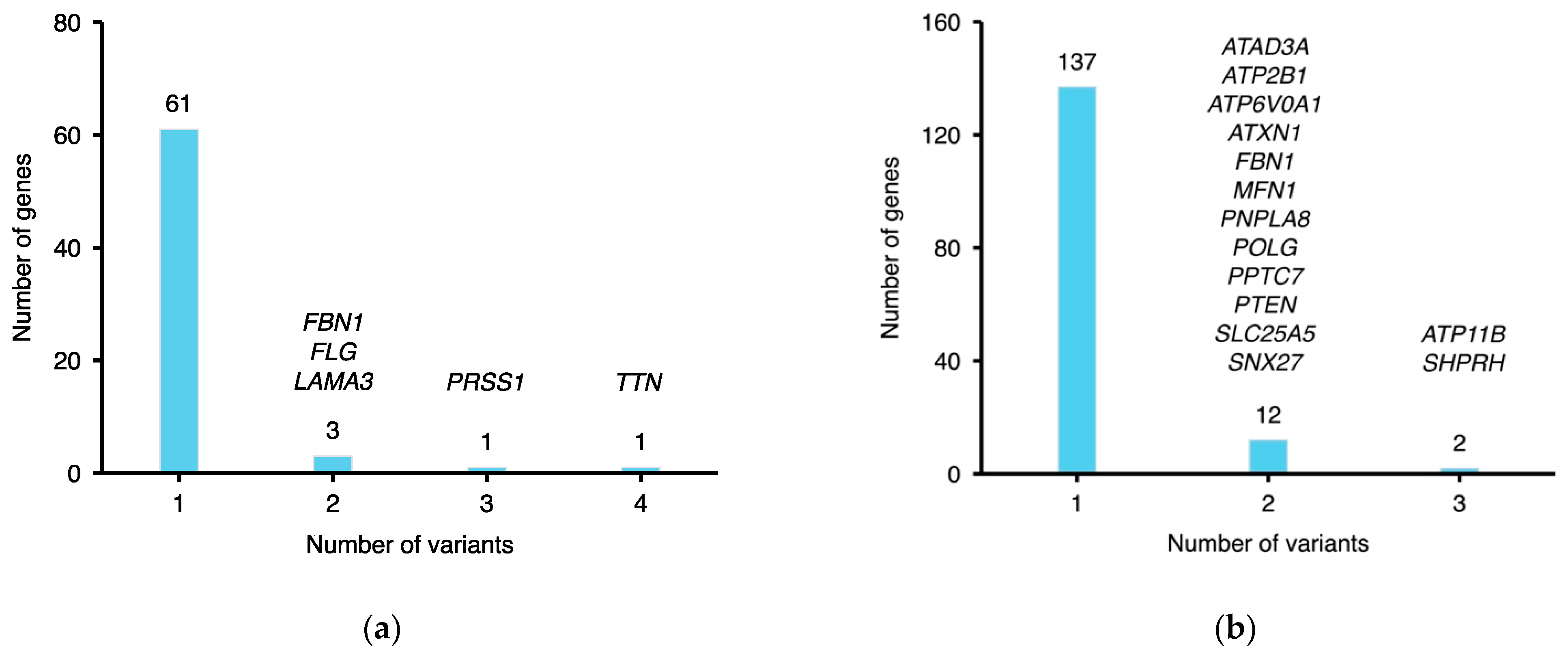
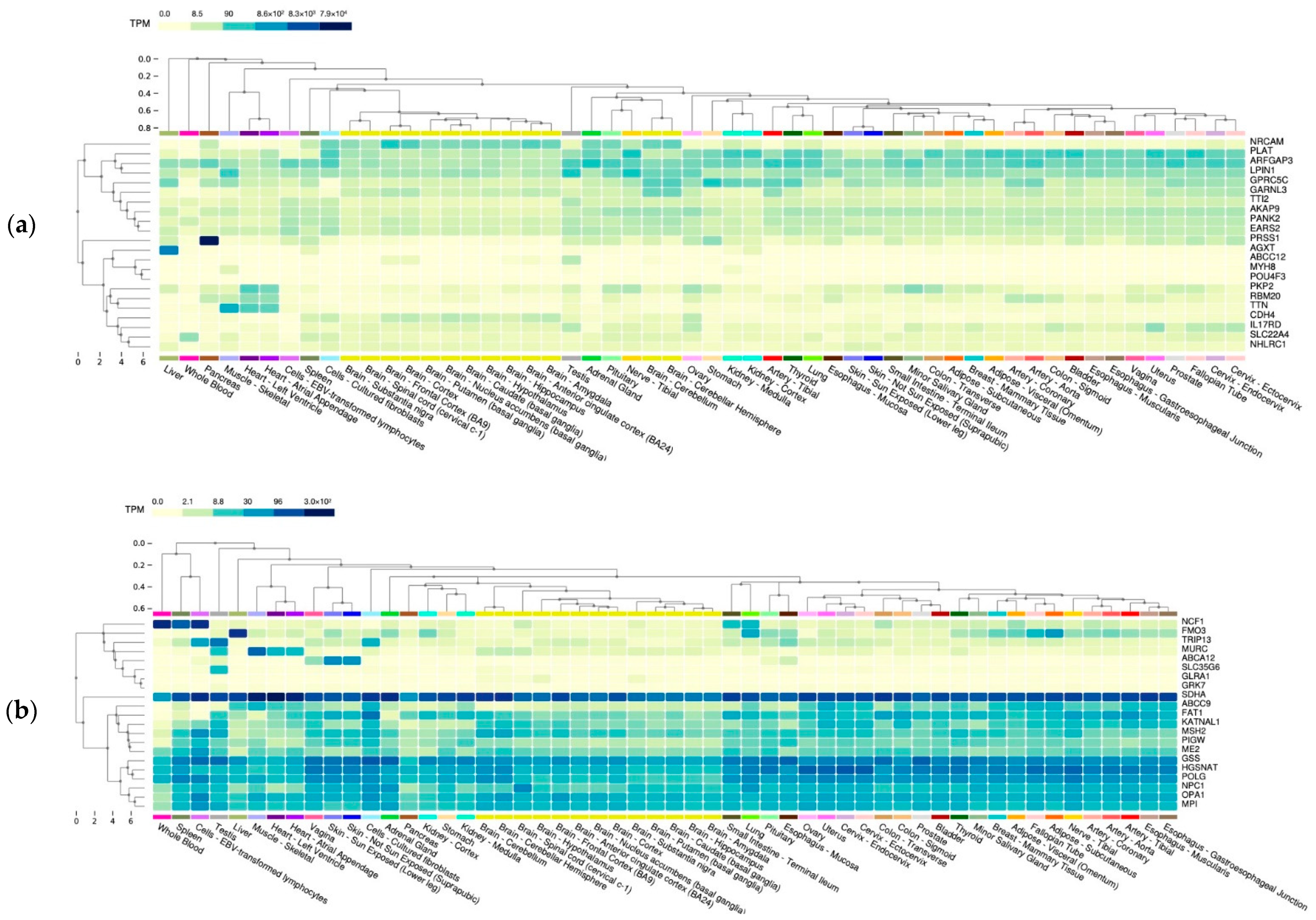
2.3. Functional Annotation of MFS Candidate Genes
2.4. Pathogenic Rare Variants on Candidate MFS Genes
2.5. The Inheritance Pattern within a Taiwanese MFS Family Cohort
2.6. Pathogenic Rare Variants Carried by Patient no.2 and no.5
3. Discussion
4. Materials and Methods
4.1. Study Subjects and Sample Preparation
4.2. Whole Exome Sequencing and Bioinformatic Analysis
4.3. Marfan Syndrome Related Genes
4.4. Functional Annotation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chiu, H.H.; Wu, M.H.; Chen, H.C.; Kao, F.Y.; Huang, S.K. Epidemiological profile of Marfan syndrome in a general population: A national database study. Mayo Clin. Proc. 2014, 89, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Demolder, A.; von Kodolitsch, Y.; Muiño-Mosquera, L.; De Backer, J. Myocardial Function, Heart Failure and Arrhythmia in Marfan Syndrome: A Systematic Literature Review. Diagnostics 2020, 10, 751. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.C. Marfan syndrome: Clinical diagnosis and management. Eur. J. Hum. Genet. 2007, 15, 724–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Bannas, P.; Rybczynski, M.; Sheikhzadeh, S.; von Kodolitsch, Y.; Derlin, T.; Yamamura, J.; Lund, G.; Adam, G.; Groth, M. Comparison of Cine-MRI and Transthoracic Echocardiography for the Assessment of Aortic Root Diameters in Patients with Suspected Marfan Syndrome. Rofo 2015, 187, 1022–1028. [Google Scholar] [CrossRef] [Green Version]
- Winther, S.; Williams, L.K.; Keir, M.; Connelly, K.A.; Bradley, T.J.; Rakowski, H.; Crean, A.M. Cardiovascular Magnetic Resonance Provides Evidence of Abnormal Myocardial Strain and Primary Cardiomyopathy in Marfan syndrome. J. Comput. Assist. Tomogr. 2019, 43, 410–415. [Google Scholar] [CrossRef]
- Radke, R.M.; Baumgartner, H. Diagnosis and treatment of Marfan syndrome: An update. Heart 2014, 100, 1382–1391. [Google Scholar] [CrossRef]
- Dietz, H.C.; Pyeritz, R.E. Mutations in the human gene for fibrillin-1 (FBN1) in the Marfan syndrome and related disorders. Hum. Mol. Genet. 1995, 4, 1799–1809. [Google Scholar] [CrossRef]
- Sakai, L.Y.; Keene, D.R.; Renard, M.; De Backer, J. FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Gene 2016, 591, 279–291. [Google Scholar] [CrossRef]
- Habashi, J.P.; Judge, D.P.; Holm, T.M.; Cohn, R.D.; Loeys, B.L.; Cooper, T.K.; Myers, L.; Klein, E.C.; Liu, G.; Calvi, C.; et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006, 312, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.K.; Rommel, K.; Mishra, A.; Karck, M.; Haverich, A.; Schmidtke, J.; Arslan-Kirchner, M. TGFBR1 and TGFBR2 mutations in patients with features of Marfan syndrome and Loeys-Dietz syndrome. Hum. Mutat. 2006, 27, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Salchow, D.J.; Gehle, P. Ocular manifestations of Marfan syndrome in children and adolescents. Eur. J. Ophthalmol. 2019, 29, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Kurup, S.P.; Fawzi, A.A.; Durbin, M.K.; Maumenee, I.H.; Mets, M.B. Comparative data on SD-OCT for the retinal nerve fiber layer and retinal macular thickness in a large cohort with Marfan syndrome. Ophthalm. Genet. 2017, 38, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Fortune, B.; Cull, G.; Reynaud, J.; Wang, L.; Burgoyne, C.F. Relating Retinal Ganglion Cell Function and Retinal Nerve Fiber Layer (RNFL) Retardance to Progressive Loss of RNFL Thickness and Optic Nerve Axons in Experimental Glaucoma. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3936–3944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackman, P.; Vihola, A.; Haravuori, H.; Marchand, S.; Sarparanta, J.; De Seze, J.; Labeit, S.; Witt, C.; Peltonen, L.; Richard, I.; et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am. J. Hum. Genet. 2002, 71, 492–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerull, B.; Gramlich, M.; Atherton, J.; McNabb, M.; Trombitás, K.; Sasse-Klaassen, S.; Seidman, J.G.; Seidman, C.; Granzier, H.; Labeit, S.; et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002, 30, 201–204. [Google Scholar] [CrossRef]
- Godfrey, C.; Clement, E.; Mein, R.; Brockington, M.; Smith, J.; Talim, B.; Straub, V.; Robb, S.; Quinlivan, R.; Feng, L.; et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007, 130, 2725–2735. [Google Scholar] [CrossRef] [Green Version]
- Attanasio, M.; Lapini, I.; Evangelisti, L.; Lucarini, L.; Giusti, B.; Porciani, M.; Fattori, R.; Anichini, C.; Abbate, R.; Gensini, G.; et al. FBN1 mutation screening of patients with Marfan syndrome and related disorders: Detection of 46 novel FBN1 mutations. Clin. Genet. 2008, 74, 39–46. [Google Scholar] [CrossRef]
- Fang, M.; Yu, C.; Chen, S.; Xiong, W.; Li, X.; Zeng, R.; Zhuang, J.; Fan, R. Identification of Novel Clinically Relevant Variants in 70 Southern Chinese patients with Thoracic Aortic Aneurysm and Dissection by Next-generation Sequencing. Sci. Rep. 2017, 7, 10035. [Google Scholar] [CrossRef]
- Feldmann, J.; Callebaut, I.; Raposo, G.; Certain, S.; Bacq, D.; Dumont, C.; Lambert, N.; Ouachée-Chardin, M.; Chedeville, G.; Tamary, H.; et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 2003, 115, 461–473. [Google Scholar] [CrossRef] [Green Version]
- Kubota, Y.; Nomura, K.; Katoh, Y.; Yamashita, R.; Kaneko, K.; Furuyama, K. Novel Mechanisms for Heme-dependent Degradation of ALAS1 Protein as a Component of Negative Feedback Regulation of Heme Biosynthesis. J. Biol. Chem. 2016, 291, 20516–20529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Wijk, K.; Akabane, T.; Kimura, T.; Saitoh, S.; Okano, S.; Kelly, V.P.; Takagi, M.; Kodama, K.; Takahashi, K.; Tanaka, T.; et al. Heterozygous disruption of ALAS1 in mice causes an accelerated age-dependent reduction in free heme, but not total heme, in skeletal muscle and liver. Arch Biochem. Biophys. 2021, 697, 108721. [Google Scholar] [CrossRef] [PubMed]
- Sijia, L.; Shuangxin, L.; Wei, S.; Yanhai, C. Marfan syndrome with antineutrophil cytoplasmic antibody-associated systemic vasculitis presenting as severe anaemia and haematuria after the Bentall procedure. Eur. J. Cardiothorac. Surg. 2013, 44, 379–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tharp, C.A.; Haywood, M.E.; Sbaizero, O.; Taylor, M.R.G.; Mestroni, L. The Giant Protein Titin’s Role in Cardiomyopathy: Genetic, Transcriptional, and Post-translational Modifications of TTN and Their Contribution to Cardiac Disease. Front. Physiol. 2019, 10, 1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bello, L.; Melacini, P.; Pezzani, R.; D’Amico, A.; Piva, L.; Leonardi, E.; Torella, A.; Soraru, G.; Palmieri, A.; Smaniotto, G.; et al. Cardiomyopathy in patients with POMT1-related congenital and limb-girdle muscular dystrophy. Eur. J. Hum. Genet. 2012, 20, 1234–1239. [Google Scholar] [CrossRef] [Green Version]
- Novelli, V.; Malkani, K.; Cerrone, M. Pleiotropic Phenotypes Associated With PKP2 Variants. Front. Cardiovasc. Med. 2018, 5, 184. [Google Scholar] [CrossRef]
- Zeharia, A.; Shaag, A.; Houtkooper, R.H.; Hindi, T.; de Lonlay, P.; Erez, G.; Hubert, L.; Saada, A.; de Keyzer, Y.; Eshel, G.; et al. Mutations in LPIN1 cause recurrent acute myoglobinuria in childhood. Am. J. Hum. Genet. 2008, 83, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef]
- Ogata, T.; Ueyama, T.; Isodono, K.; Tagawa, M.; Takehara, N.; Kawashima, T.; Harada, K.; Takahashi, T.; Shioi, T.; Matsubara, H.; et al. MURC, a muscle-restricted coiled-coil protein that modulates the Rho/ROCK pathway, induces cardiac dysfunction and conduction disturbance. Mol. Cell Biol. 2008, 28, 3424–3436. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, N.; Ogata, T.; Naito, D.; Miyagawa, K.; Taniguchi, T.; Hamaoka, T.; Maruyama, N.; Kasahara, T.; Nishi, M.; Matoba, S.; et al. MURC deficiency in smooth muscle attenuates pulmonary hypertension. Nat. Commun. 2016, 7, 12417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, G.; Ueyama, T.; Ogata, T.; Czernuszewicz, G.; Tan, Y.; Dorn, G.W., 2nd; Bogaev, R.; Amano, K.; Oh, H.; Matsubara, H.; et al. Molecular genetic and functional characterization implicate muscle-restricted coiled-coil gene (MURC) as a causal gene for familial dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2011, 4, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.Y.; Yang, J.H.; Yeh, E.C.; Tsai, M.F.; Kao, H.J.; Lo, C.Z.; Chang, L.P.; Lin, W.J.; Hsieh, F.J.; Belsare, S.; et al. Genetic profiles of 103,106 individuals in the Taiwan Biobank provide insights into the health and history of Han Chinese. NPJ Genom Med. 2021, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [Green Version]
- Fatini, C.; Attanasio, M.; Porciani, C.; Sticchi, E.; Padeletti, L.; Lapini, I.; Abbate, R.; Gensini, G.F.; Pepe, G. AGT and ACE genes influence classic mitral valve prolapse predisposition in Marfan patients. Int. J. Cardiol. 2008, 123, 293–297. [Google Scholar] [CrossRef]
- Gentilini, D.; Oliveri, A.; Fazia, T.; Pini, A.; Marelli, S.; Bernardinelli, L.; Di Blasio, A.M. NGS analysis in Marfan syndrome spectrum: Combination of rare and common genetic variants to improve genotype-phenotype correlation analysis. PLoS ONE 2019, 14, e0222506. [Google Scholar] [CrossRef]
- Lerner-Ellis, J.P.; Aldubayan, S.H.; Hernandez, A.L.; Kelly, M.A.; Stuenkel, A.J.; Walsh, J.; Joshi, V.A. The spectrum of FBN1, TGFβR1, TGFβR2 and ACTA2 variants in 594 individuals with suspected Marfan Syndrome, Loeys-Dietz Syndrome or Thoracic Aortic Aneurysms and Dissections (TAAD). Mol. Genet. Metab. 2014, 112, 171–176. [Google Scholar] [CrossRef]
- Ritelli, M.; Dordoni, C.; Venturini, M.; Chiarelli, N.; Quinzani, S.; Traversa, M.; Zoppi, N.; Vascellaro, A.; Wischmeijer, A.; Manfredini, E.; et al. Clinical and molecular characterization of 40 patients with classic Ehlers-Danlos syndrome: Identification of 18 COL5A1 and 2 COL5A2 novel mutations. Orphanet. J. Rare Dis. 2013, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Gan, Q.; Liu, Q.; Hu, X.; You, C. Collagen Type I Alpha 2 (COL1A2) Polymorphism Contributes to Intracranial Aneurysm Susceptibility: A Meta-Analysis. Med. Sci. Monit. 2017, 23, 3240–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenstrup, R.J.; Florer, J.B.; Willing, M.C.; Giunta, C.; Steinmann, B.; Young, F.; Susic, M.; Cole, W.G. COL5A1 haploinsufficiency is a common molecular mechanism underlying the classical form of EDS. Am. J. Hum. Genet. 2000, 66, 1766–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Yu, J.; Wang, K.; Wang, B.; Wang, M.; Zhang, S.; Qin, S.; Yu, Z. Exome sequencing identified new mutations in a Marfan syndrome family. Diagn. Pathol. 2014, 9, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Lu, C.; Wu, W.; Liu, Y.; Wang, R.; Si, N.; Meng, X.; Zhang, S.; Zhang, X. Application of next-generation sequencing to screen for pathogenic mutations in 123 unrelated Chinese patients with Marfan syndrome or a related disease. Sci. China Life Sci. 2019, 62, 1630–1637. [Google Scholar] [CrossRef]
- Tsipouras, P.; Del Mastro, R.; Sarfarazi, M.; Lee, B.; Vitale, E.; Child, A.H.; Godfrey, M.; Devereux, R.B.; Hewett, D.; Steinmann, B.; et al. Genetic linkage of the Marfan syndrome, ectopia lentis, and congenital contractural arachnodactyly to the fibrillin genes on chromosomes 15 and The International Marfan Syndrome Collaborative Study. N. Engl. J. Med. 1992, 326, 905–909. [Google Scholar] [CrossRef]
- Uyeda, T.; Takahashi, T.; Eto, S.; Sato, T.; Xu, G.; Kanezaki, R.; Toki, T.; Yonesaka, S.; Ito, E. Three novel mutations of the fibrillin-1 gene and ten single nucleotide polymorphisms of the fibrillin-3 gene in Marfan syndrome patients. J. Hum. Genet. 2004, 49, 404–407. [Google Scholar] [CrossRef] [Green Version]
- Meester, J.A.; Vandeweyer, G.; Pintelon, I.; Lammens, M.; Van Hoorick, L.; De Belder, S.; Waitzman, K.; Young, L.; Markham, L.W.; Vogt, J.; et al. Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet. Med. 2017, 19, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Busnadiego, O.; Gorbenko Del Blanco, D.; González-Santamaría, J.; Habashi, J.P.; Calderon, J.F.; Sandoval, P.; Bedja, D.; Guinea-Viniegra, J.; Lopez-Cabrera, M.; Rosell-Garcia, T.; et al. Elevated expression levels of lysyl oxidases protect against aortic aneurysm progression in Marfan syndrome. J. Mol. Cell Cardiol. 2015, 85, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Loeys, B.L.; Mortier, G.; Dietz, H.C. Bone lessons from Marfan syndrome and related disorders: Fibrillin, TGF-B and BMP at the balance of too long and too short. Pediatr. Endocrinol. Rev. 2013, 10 (Suppl. 2), 417–423. [Google Scholar]
- Morlino, S.; Alesi, V.; Calì, F.; Lepri, F.R.; Secinaro, A.; Grammatico, P.; Novelli, A.; Drago, F.; Castori, M.; Baban, A. LTBP2-related "Marfan-like" phenotype in two Roma/Gypsy subjects with the LTBP2 homozygous p.R299X variant. Am. J. Med. Genet. A 2019, 179, 104–112. [Google Scholar] [CrossRef]
- Giusti, B.; Porciani, M.C.; Brunelli, T.; Evangelisti, L.; Fedi, S.; Gensini, G.F.; Abbate, R.; Sani, G.; Yacoub, M.; Pepe, G. Phenotypic variability of cardiovascular manifestations in Marfan Syndrome. Possible role of hyperhomocysteinemia and C677T MTHFR gene polymorphism. Eur. Heart J. 2003, 24, 2038–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, N.; Komuro, I. Genetic basis of hereditary thoracic aortic aneurysms and dissections. J. Cardiol. 2019, 74, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Cannaerts, E.; Kempers, M.; Maugeri, A.; Marcelis, C.; Gardeitchik, T.; Richer, J.; Micha, D.; Beauchesne, L.; Timmermans, J.; Vermeersch, P.; et al. Novel pathogenic SMAD2 variants in five families with arterial aneurysm and dissection: Further delineation of the phenotype. J. Med. Genet. 2019, 56, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Holm, T.M.; Habashi, J.P.; Doyle, J.J.; Bedja, D.; Chen, Y.; van Erp, C.; Lindsay, M.E.; Kim, D.; Schoenhoff, F.; Cohn, R.D.; et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 2011, 332, 358–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourquin, J.; Duncan, D.; Shi, Z.; Zhang, B. GLAD4U: Deriving and prioritizing gene lists from PubMed literature. BMC Genom. 2012, 13 (Suppl. 8), S20. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucl. Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [Green Version]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.L.; Goldsmith, C.A.; Eppig, J.T. The Mammalian Phenotype Ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 2005, 6, R7. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology Consortium, The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Gender | Age | Relatedness | Note | Systemic Score |
|---|---|---|---|---|---|
| pt.1 | F | 52 | - | - | 9 |
| pt.2 | F | 42 | - | - | 5 |
| pt.3 | F | 40 | - | - | 4 |
| pt.4 | M | 43 | - | - | 7 |
| pt.5 | M | 42 | - | - | 7 |
| pt.6 | M | 37 | - | - | 4 |
| pt.7 | F | 52 | Family 1-Mother | - | 4 |
| pt.8 | M | 23 | Family 1-Son-01 | - | 6 |
| pt.9 | M | 26 | Family 1-Son-02 | - | 4 |
| pt.10 | M | - | Family 1-Father | Healthy control | - |
| pt.11 | F | 20 | Family 1-Daughter | - | 7 |
| Chr. | Position | Ref. | Alt. | Gene | Type | Number of Alt. Allele in MFS Patients | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 11 | 10 b | ||||||
| 1 | 11794020 | G | A | MTHFR | nsSNV | 1 | - | - | - | - | - | - | - | - | - | - |
| 2 | 189051324 | T | C | COL5A2 | nsSNV | 1 | - | - | - | - | - | - | - | - | - | - |
| 3 | 30623240 | A | C | TGFBR2 | nsSNV | - | - | - | 1 | - | - | - | - | - | - | - |
| 9 | 99105216 | C | T | TGFBR1 | nsSNV | - | - | - | - | - | - | - | - | - | - | 1 |
| 9 | 134758250 | G | A | COL5A1 | nsSNV | - | - | - | - | - | 1 | - | - | - | - | - |
| 11 | 65557857 | CCAG | CCAGCAGCAG,C | LTBP3 | Ins (NF) | - | 1 | - | - | - | 1 a | - | - | - | - | - |
| 12 | 57167041 | C | T | LRP1 | nsSNV | - | - | - | - | - | 1 | - | - | - | - | - |
| 15 | 48420690 | C | T | FBN1 | nsSNV | 1 | - | - | - | - | - | - | - | - | - | - |
| 15 | 48436991 | C | A | FBN1 | nsSNV | - | - | - | - | - | - | 1 | 1 | 1 | 1 | - |
| 15 | 48437824 | G | T | FBN1 | nsSNV | - | - | - | 1 | - | - | - | - | - | - | - |
| 15 | 48448860 | C | T | FBN1 | nsSNV | - | - | 1 | - | - | - | - | - | - | - | - |
| 15 | 48487321 | GC | G | FBN1 | Del (F) | - | - | - | - | - | 1 | - | - | - | - | - |
| 15 | 48503803 | GAC | G | FBN1 | Del (F) | 1 | - | - | - | - | - | - | - | - | - | - |
| 16 | 15714999 | T | C | MYH11 | nsSNV | - | - | - | - | - | - | 1 | - | 1 | - | - |
| 17 | 63494011 | G | T | ACE | nsSNV | - | - | 1 | - | - | - | - | - | - | - | - |
| 17 | 63497343 | G | T | ACE | stop gain | - | - | 1 | - | - | - | - | - | - | - | - |
| 19 | 8123973 | C | T | FBN3 | nsSNV | - | - | 1 | - | - | - | - | - | - | - | - |
| 19 | 8133099 | CGTT | C | FBN3 | Del (NF) | - | - | - | - | - | - | - | - | - | - | 1 |
| 19 | 8136499 | A | G | FBN3 | nsSNV | - | - | 1 | - | - | - | - | - | - | - | - |
| Functional Annotations | No. of Reference Genes in the Category | No. of MFS Candidate Genes in the Category | p-Value | q-Value (FDR) | |
|---|---|---|---|---|---|
| Knockout Mouse Phenotype Category | |||||
| Abnormal cardiovascular system morphology | 1794 | 55 | 1.40 × 10−7 | 9.11 × 10−4 | |
| Abnormal sarcomere morphology | 60 | 9 | 3.13 × 10−7 | 1.02 × 10−3 | |
| Premature death | 952 | 34 | 2.80 × 10−6 | 6.09 × 10−3 | |
| Kyphosis | 162 | 12 | 7.80 × 10−6 | 1.27 × 10−2 | |
| Abnormal heart morphology | 1297 | 40 | 1.17 × 10−5 | 1.49 × 10−2 | |
| Abnormal blood vessel morphology | 1070 | 35 | 1.38 × 10−5 | 1.49 × 10−2 | |
| Abnormal stria vascularis morphology | 40 | 6 | 3.16 × 10−5 | 2.92 × 10−2 | |
| Ascending aorta aneurysm | 5 | 3 | 3.59 × 10−5 | 2.92 × 10−2 | |
| GO term Category | |||||
| Sensory perception of light stimulus | 209 | 14 | 7.25 × 10−7 | 9.45 × 10−4 | |
| ATPase activity | 438 | 18 | 2.09 × 10−5 | 1.01 × 10−2 | |
| Extracellular structure organization | 400 | 17 | 2.33 × 10−5 | 1.01 × 10−2 | |
| Contractile fiber | 226 | 12 | 4.70 × 10−5 | 1.53 × 10−2 | |
| Sensory system development | 355 | 15 | 7.60 × 10−5 | 1.98 × 10−2 | |
| Extracellular matrix | 496 | 18 | 1.04 × 10−4 | 2.27 × 10−2 | |
| Actinin binding | 39 | 5 | 1.52 × 10−4 | 2.83 × 10−2 | |
| Extracellular matrix structural constituent | 158 | 9 | 2.50 × 10−4 | 3.54 × 10−2 | |
| Vacuolar membrane | 397 | 15 | 2.58 × 10−4 | 3.54 × 10−2 | |
| Structural constituent of muscle | 44 | 5 | 2.71 × 10−4 | 3.54 × 10−2 | |
| Organic hydroxy compound metabolic process | 500 | 17 | 3.49 × 10−4 | 3.93 × 10−2 | |
| Urogenital system development | 326 | 13 | 4.00 × 10−4 | 3.93 × 10−2 | |
| Cell junction organization | 285 | 12 | 4.11 × 10−4 | 3.93 × 10−2 | |
| Multicellular organismal signaling | 170 | 9 | 4.29 × 10−4 | 3.93 × 10−2 | |
| Protein kinase C binding | 49 | 5 | 4.52 × 10−4 | 3.93 × 10−2 | |
| Chr. | Position | Ref. | Alt. | Gene | Type | avSNP150 | Allelic Frequency | Pathogenicity | MFS a | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1KGP | gnomAD | ExAC | TWB | ClinVar | CADD | REVEL | ||||||||
| (EAS) | (EAS) | (EAS) | ||||||||||||
| 9 | 131513282 | G | A | POMT1 | nsSNV | rs146869947 | 0.001 | 0.0019 | 0.0027 | 0.003 | Conflicting interpretations of pathogenicity | 26.9 | 0.836 | 7, 9, 11 |
| 15 | 48448860 | C | T | FBN1 | nsSNV | - | - | - | - | - | Pathogenic | 32 | 0.988 | 3 |
| 17 | 75831135 | C | T | UNC13D | nsSNV | rs140184929 | 0.006 | 0.0054 | 0.0043 | 0.003 | Conflicting interpretations of pathogenicity | 27.9 | 0.933 | 4 |
| Chr. | Position | Ref. | Alt. | Gene | Type | Allelic Frequency | Alt. Allele of | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| a MFS Family | |||||||||||
| TWB | 7 | 8 | 9 | 11 | 10 a | ||||||
| 2 | 178794954 | C | T | TTN | nsSNV | 0.007 | 1 | - | 1 | 1 | - |
| 3 | 52199279 | G | A | ALAS1 | nsSNV | 0.0005 | 1 | 1 | 1 | 1 | - |
| 3 | 107716694 | A | G | BBX | nsSNV | - | 1 | 1 | - | 1 | - |
| 9 | 131513282 | G | A | POMT1 | nsSNV | 0.003 | 1 | - | 1 | 1 | - |
| 12 | 109264325 | C | T | ACACB | nsSNV | - | 1 | 1 | - | 1 | - |
| 15 | 40856938 | A | G | SPINT1 | nsSNV | - | 1 | - | 1 | 1 | - |
| 15 | 48436991 | C | A | FBN1 # | nsSNV | - | 1 | 1 | 1 | 1 | - |
| 20 | 45895081 | G | A | CTSA | nsSNV | 0.004 | 1 | 1 | 1 | - | - |
| 20 | 63350423 | C | T | CHRNA4 | nsSNV | - | 1 | - | 1 | 1 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, M.-R.; Chang, C.-M.; Ting, J.; Chang, J.-G.; Chou, W.-H.; Huang, K.-J.; Cheng, G.; Chang, H.-H.; Chang, W.-C. Application of Whole Exome Sequencing and Functional Annotations to Identify Genetic Variants Associated with Marfan Syndrome. J. Pers. Med. 2022, 12, 198. https://doi.org/10.3390/jpm12020198
Lin M-R, Chang C-M, Ting J, Chang J-G, Chou W-H, Huang K-J, Cheng G, Chang H-H, Chang W-C. Application of Whole Exome Sequencing and Functional Annotations to Identify Genetic Variants Associated with Marfan Syndrome. Journal of Personalized Medicine. 2022; 12(2):198. https://doi.org/10.3390/jpm12020198
Chicago/Turabian StyleLin, Min-Rou, Che-Mai Chang, Jafit Ting, Jan-Gowth Chang, Wan-Hsuan Chou, Kuei-Jung Huang, Gloria Cheng, Hsiao-Huang Chang, and Wei-Chiao Chang. 2022. "Application of Whole Exome Sequencing and Functional Annotations to Identify Genetic Variants Associated with Marfan Syndrome" Journal of Personalized Medicine 12, no. 2: 198. https://doi.org/10.3390/jpm12020198