
Biobanking as a Tool for Genomic Research: From Allele Frequencies to Cross-Ancestry Association Studies
 ,
,  , , and
, , and 
Abstract
:1. Introduction
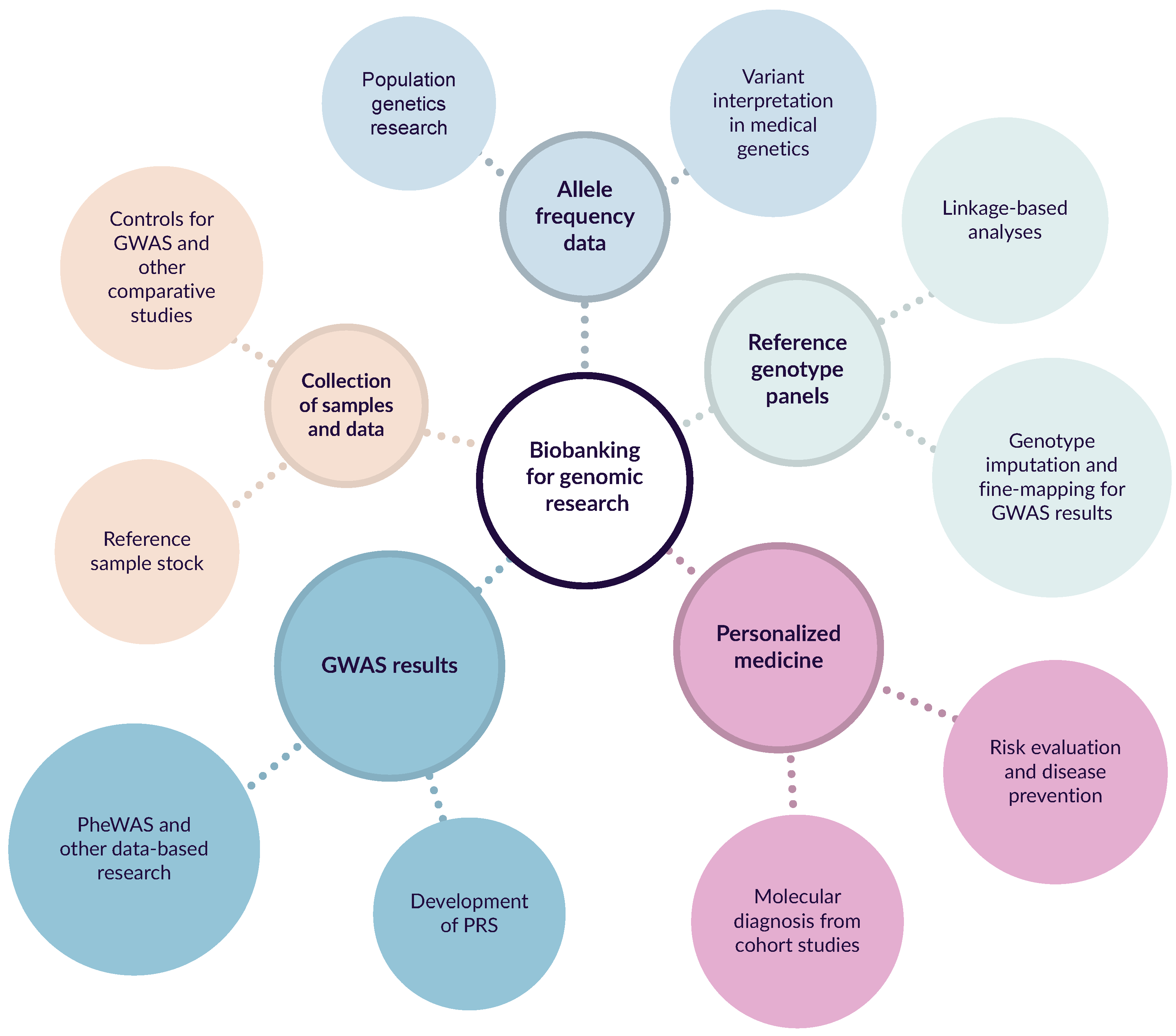
2. Applications of Biobanking in Genomic Medicine and Research
3. World’s Largest Biobanks and Genomic Research
3.1. UK Biobank (UKB)
3.2. BioBank Japan (BBJ)
3.3. FinnGen Research Project
3.4. Estonian Biobank (EB)
3.5. China Kadoorie Biobank (CKB)
3.6. Tohoku Medical Megabank Project (TMM)
3.7. Taiwan Biobank (TWB)
3.8. LifeLines Cohort Study
3.9. Other Biobanks
4. Data Integration and Prospects of Trans-Biobank Research
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GWAS | Genome-wide association study |
| SNP | single-nucleotide polymorphism |
| UKB | UK Biobank |
| BBJ | BioBank Japan |
| EB | Estonian biobank |
| CKB | China Kadoorie Biobank |
| TMM | Tohoku Medical Megabank Project |
| WES | whole-exome sequencing |
| WGS | whole-genome sequencing |
References
- Parodi, B. Biobanks: A Definition. In Ethics, Law and Governance of Biobanking; Springer: Dordrecht, The Netherlands, 2015; pp. 15–19. [Google Scholar] [CrossRef]
- Cambon-Thomsen, A.; Ducournau, P.; Gourraud, P.A.; Pontille, D. Biobanks for Genomics and Genomics for Biobanks. Comp. Funct. Genom. 2003, 4, 628–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Daw Elbait, G.; Henschel, A.; Tay, G.K.; Al Safar, H.S. A Population-Specific Major Allele Reference Genome From The United Arab Emirates Population. Front. Genet. 2021, 12, 428. [Google Scholar] [CrossRef] [PubMed]
- Takayama, J.; Tadaka, S.; Yano, K.; Katsuoka, F.; Gocho, C.; Funayama, T.; Makino, S.; Okamura, Y.; Kikuchi, A.; Sugimoto, S.; et al. Construction and integration of three de novo Japanese human genome assemblies toward a population-specific reference. Nat. Commun. 2021, 12, 226. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, D.I.; Wijmenga, C.; Slagboom, E.P.; Swertz, M.A.; Karssen, L.C.; Abdellaoui, A.; Ye, K.; Guryev, V.; Vermaat, M.; van Dijk, F.; et al. The Genome of the Netherlands: Design, and project goals. Eur. J. Hum. Genet. 2014, 22, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Barbitoff, Y.A.; Skitchenko, R.K.; Poleshchuk, O.I.; Shikov, A.E.; Serebryakova, E.A.; Nasykhova, Y.A.; Polev, D.E.; Shuvalova, A.R.; Shcherbakova, I.V.; Fedyakov, M.A.; et al. Whole-exome sequencing provides insights into monogenic disease prevalence in Northwest Russia. Mol. Genet. Genom. Med. 2019, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Barbitoff, Y.A.; Khmelkova, D.N.; Pomerantseva, E.A.; Slepchenkov, A.V.; Zubashenko, N.A.; Mironova, I.V.; Kaimonov, V.S.; Polev, D.E.; Tsay, V.V.; Glotov, A.S.; et al. Expanding the Russian allele frequency reference via cross-laboratory data integration: Insights from 6,096 exome samples. medRxiv 2021. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Samocha, K.E.; Banks, E.; Fennell, T.; O, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; Birnbaum, D.P.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 283–291. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Reich, D.; Thangaraj, K.; Patterson, N.; Price, A.L.; Singh, L. Reconstructing Indian population history. Nature 2009, 461, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Cassa, C.A.; Weghorn, D.; Balick, D.J.; Jordan, D.M.; Nusinow, D.; Samocha, K.E.; O’Donnell-Luria, A.; MacArthur, D.G.; Daly, M.J.; Beier, D.R.; et al. Estimating the selective effects of heterozygous protein-truncating variants from human exome data. Nat. Genet. 2017, 49, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Havrilla, J.M.; Pedersen, B.S.; Layer, R.M.; Quinlan, A.R. A map of constrained coding regions in the human genome. Nat. Genet. 2019, 51, 88–95. [Google Scholar] [CrossRef]
- Halldorsson, B.V.; Eggertsson, H.P.; Moore, K.H.S.; Hauswedell, H.; Eiriksson, O.; Ulfarsson, M.O.; Palsson, G.; Hardarson, M.T.; Oddsson, A.; Jensson, B.O.; et al. The sequences of 150,119 genomes in the UK biobank. bioRxiv 2022. [Google Scholar] [CrossRef]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [Green Version]
- Schaid, D.J.; Chen, W.; Larson, N.B. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 2018, 19, 491–504. [Google Scholar] [CrossRef]
- Nielsen, R.; Hellmann, I.; Hubisz, M.; Bustamante, C.; Clark, A.G. Recent and ongoing selection in the human genome. Nat. Rev. Genet. 2007, 8, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Loh, P.R.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; A Reshef, Y.; K Finucane, H.; Schoenherr, S.; Forer, L.; McCarthy, S.; Abecasis, G.R.; et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet 2016, 48, 1443–1448. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lee, S. Integrating external controls in case–control studies improves power for rare-variant tests. Genet. Epidemiol. 2022, 46, 145–158. [Google Scholar] [CrossRef]
- Sanderson, E.; Glymour, M.M.; Holmes, M.V.; Kang, H.; Morrison, J.; Munafò, M.R.; Palmer, T.; Schooling, C.M.; Wallace, C.; Zhao, Q.; et al. Mendelian randomization. Nat. Rev. Methods Prim. 2022, 2, 6. [Google Scholar] [CrossRef]
- Mills, M.C.; Rahal, C. A scientometric review of genome-wide association studies. Commun. Biol. 2019, 2, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Chen, Y.; Ritchie, M.D.; Moore, J.H. Electronic health records and polygenic risk scores for predicting disease risk. Nat. Rev. Genet. 2020, 21, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Treff, N.R.; Marin, D.; Lello, L.; Hsu, S.; Tellier, L.C.A.M. PREIMPLANTATION GENETIC TESTING: Preimplantation genetic testing for polygenic disease risk. Reproduction 2020, 160, A13–A17. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Stringer, S.; Frei, O.; Umićević Mirkov, M.; de Leeuw, C.; Polderman, T.J.C.; van der Sluis, S.; Andreassen, O.A.; Neale, B.M.; Posthuma, D. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 2019, 51, 1339–1348. [Google Scholar] [CrossRef]
- Shikov, A.E.; Skitchenko, R.K.; Predeus, A.V.; Barbitoff, Y.A. Phenome-wide functional dissection of pleiotropic effects highlights key molecular pathways for human complex traits. Sci. Rep. 2020, 10, 1037. [Google Scholar] [CrossRef] [Green Version]
- Kawame, H.; Fukushima, A.; Fuse, N.; Nagami, F.; Suzuki, Y.; Sakurai-Yageta, M.; Yasuda, J.; Yamaguchi-Kabata, Y.; Kinoshita, K.; Ogishima, S.; et al. The return of individual genomic results to research participants: Design and pilot study of Tohoku Medical Megabank Project. J. Hum. Genet. 2022, 67, 9–17. [Google Scholar] [CrossRef]
- Alver, M.; Palover, M.; Saar, A.; Läll, K.; Zekavat, S.M.; Tõnisson, N.; Leitsalu, L.; Reigo, A.; Nikopensius, T.; Ainla, T.; et al. Recall by genotype and cascade screening for familial hypercholesterolemia in a population-based biobank from Estonia. Genet. Med. 2019, 21, 1173–1180. [Google Scholar] [CrossRef] [Green Version]
- All of Us Research Program Investigators; Denny, J.C.; Rutter, J.L.; Goldstein, D.B.; Philippakis, A.; Smoller, J.W.; Jenkins, G.; Dishman, E. The “All of Us” Research Program. N. Engl. J. Med. 2019, 381, 668–676. [Google Scholar] [CrossRef]
- Federici, G.; Soddu, S. Variants of uncertain significance in the era of high-throughput genome sequencing: A lesson from breast and ovary cancers. J. Exp. Clin. Cancer Res. 2020, 39, 46. [Google Scholar] [CrossRef] [Green Version]
- Vears, D.F.; Niemiec, E.; Howard, H.C.; Borry, P. Analysis of VUS reporting, variant reinterpretation and recontact policies in clinical genomic sequencing consent forms. Eur. J. Hum. Genet. 2018, 26, 1743–1751. [Google Scholar] [CrossRef] [Green Version]
- Schoot, V.v.d.; Viellevoije, S.J.; Tammer, F.; Brunner, H.G.; Arens, Y.; Yntema, H.G.; Oerlemans, A.J.M. The impact of unsolicited findings in clinical exome sequencing, a qualitative interview study. Eur. J. Hum. Genet 2021, 29, 930–939. [Google Scholar] [CrossRef]
- De Wert, G.; Dondorp, W.; Clarke, A.; Dequeker, E.M.C.; Cordier, C.; Deans, Z.; van El, C.G.; Fellmann, F.; Hastings, R.; Hentze, S.; et al. Opportunistic genomic screening. Recommendations of the European Society of Human Genetics. Eur. J. Hum. Genet. 2021, 29, 365–377. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Gordon, A.S.; Amendola, L.M.; Adelman, K.; Bale, S.J.; Chung, W.K.; Gollob, M.H.; Harrison, S.M.; Herman, G.E.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1391–1398. [Google Scholar] [CrossRef]
- O’Donoghue, S.; Dee, S.; Byrne, J.A.; Watson, P.H. How Many Health Research Biobanks Are There? Biopreservation Biobanking 2022, 20, 224–228. [Google Scholar] [CrossRef]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Wells, H.R.; Freidin, M.B.; Zainul Abidin, F.N.; Payton, A.; Dawes, P.; Munro, K.J.; Morton, C.C.; Moore, D.R.; Dawson, S.J.; Williams, F.M. GWAS Identifies 44 Independent Associated Genomic Loci for Self-Reported Adult Hearing Difficulty in UK Biobank. Am. J. Hum. Genet. 2019, 105, 788–802. [Google Scholar] [CrossRef] [Green Version]
- Van Hout, C.V.; Tachmazidou, I.; Backman, J.D.; Hoffman, J.D.; Liu, D.; Pandey, A.K.; Gonzaga-Jauregui, C.; Khalid, S.; Ye, B.; Banerjee, N.; et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 2020, 586, 749–756. [Google Scholar] [CrossRef]
- De Vincentis, A.; Tavaglione, F.; Spagnuolo, R.; Pujia, R.; Tuccinardi, D.; Mascianà, G.; Picardi, A.; Antonelli Incalzi, R.; Valenti, L.; Romeo, S.; et al. Metabolic and genetic determinants for progression to severe liver disease in subjects with obesity from the UK Biobank. Int. J. Obes. 2022, 46, 486–493. [Google Scholar] [CrossRef]
- Ishigaki, K.; Akiyama, M.; Kanai, M.; Takahashi, A.; Kawakami, E.; Sugishita, H.; Sakaue, S.; Matoba, N.; Low, S.K.; Okada, Y.; et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat. Genet. 2020, 52, 669–679. [Google Scholar] [CrossRef]
- Matoba, N.; Akiyama, M.; Ishigaki, K.; Kanai, M.; Takahashi, A.; Momozawa, Y.; Ikegawa, S.; Ikeda, M.; Iwata, N.; Hirata, M.; et al. GWAS of 165,084 Japanese individuals identified nine loci associated with dietary habits. Nat. Hum. Behav. 2020, 4, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Desch, K.C.; Ozel, A.B.; Halvorsen, M.; Jacobi, P.M.; Golden, K.; Underwood, M.; Germain, M.; Tregouet, D.A.; Reitsma, P.H.; Kearon, C.; et al. Whole-exome sequencing identifies rare variants in STAB2 associated with venous thromboembolic disease. Blood 2020, 136, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Kurki, M.I.; Karjalainen, J.; Palta, P.; Sipilä, T.P.; Kristiansson, K.; Donner, K.; Reeve, M.P.; Laivuori, H.; Aavikko, M.; Kaunisto, M.A.; et al. FinnGen: Unique genetic insights from combining isolated population and national health register data. medRxiv 2022. [Google Scholar] [CrossRef]
- Sun, B.B.; Kurki, M.I.; Foley, C.N.; Mechakra, A.; Chen, C.Y.; Marshall, E.; Wilk, J.B.; Sun, B.B.; Ghen, C.Y.; Marshall, E.; et al. Genetic associations of protein-coding variants in human disease. Nature 2022, 603, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Reisberg, S.; Krebs, K.; Lepamets, M.; Kals, M.; Mägi, R.; Metsalu, K.; Lauschke, V.M.; Vilo, J.; Milani, L. Translating genotype data of 44,000 biobank participants into clinical pharmacogenetic recommendations: Challenges and solutions. Genet. Med. 2019, 21, 1345–1354. [Google Scholar] [CrossRef] [Green Version]
- Spracklen, C.N.; Horikoshi, M.; Kim, Y.J.; Lin, K.; Bragg, F.; Moon, S.; Suzuki, K.; Tam, C.H.T.; Tabara, Y.; Kwak, S.H.; et al. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature 2020, 582, 240–245. [Google Scholar] [CrossRef]
- Giannakopoulou, O.; Lin, K.; Meng, X.; Su, M.H.; Kuo, P.H.; Peterson, R.E.; Awasthi, S.; Moscati, A.; Coleman, J.R.I.; Bass, N.; et al. The Genetic Architecture of Depression in Individuals of East Asian Ancestry. JAMA Psychiatry 2021, 78, 1258. [Google Scholar] [CrossRef]
- Zhu, Z.; Li, J.; Si, J.; Ma, B.; Shi, H.; Lv, J.; Cao, W.; Guo, Y.; Millwood, I.Y.; Walters, R.G.; et al. A large-scale genome-wide association analysis of lung function in the Chinese population identifies novel loci and highlights shared genetic aetiology with obesity. Eur. Respir. J. 2021, 58, 2100199. [Google Scholar] [CrossRef]
- Li, J.; Glover, J.D.; Zhang, H.; Peng, M.; Tan, J.; Mallick, C.B.; Hou, D.; Yang, Y.; Wu, S.; Liu, Y.; et al. Limb development genes underlie variation in human fingerprint patterns. Cell 2022, 185, 95–112.e18. [Google Scholar] [CrossRef]
- Watanabe, T.; Saito, T.; Rico, E.M.G.; Hishinuma, E.; Kumondai, M.; Maekawa, M.; Oda, A.; Saigusa, D.; Saito, S.; Yasuda, J.; et al. Functional characterization of 40 CYP2B6 allelic variants by assessing efavirenz 8-hydroxylation. Biochem. Pharmacol. 2018, 156, 420–430. [Google Scholar] [CrossRef]
- Tadaka, S.; Katsuoka, F.; Ueki, M.; Kojima, K.; Makino, S.; Saito, S.; Otsuki, A.; Gocho, C.; Sakurai-Yageta, M.; Danjoh, I.; et al. 3.5KJPNv2: An allele frequency panel of 3552 Japanese individuals including the X chromosome. Hum. Genome Var. 2019, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Tadaka, S.; Hishinuma, E.; Komaki, S.; Motoike, I.N.; Kawashima, J.; Saigusa, D.; Inoue, J.; Takayama, J.; Okamura, Y.; Aoki, Y.; et al. jMorp updates in 2020: Large enhancement of multi-omics data resources on the general Japanese population. Nucleic Acids Res. 2021, 49, D536–D544. [Google Scholar] [CrossRef]
- Ohneda, K.; Hiratsuka, M.; Kawame, H.; Nagami, F.; Suzuki, Y.; Suzuki, K.; Uruno, A.; Sakurai-Yageta, M.; Hamanaka, Y.; Taira, M.; et al. A Pilot Study for Return of Individual Pharmacogenomic Results to Population-Based Cohort Study Participants. JMA J. 2022, 5, 177–189. [Google Scholar] [CrossRef]
- Park, K.S. Two Approaches for a Genetic Analysis of Pompe Disease: A Literature Review of Patients with Pompe Disease and Analysis Based on Genomic Data from the General Population. Children 2021, 8, 601. [Google Scholar] [CrossRef]
- Wei, C.Y.; Yang, J.H.; Yeh, E.C.; Tsai, M.F.; Kao, H.J.; Lo, C.Z.; Chang, L.P.; Lin, W.J.; Hsieh, F.J.; Belsare, S.; et al. Genetic profiles of 103,106 individuals in the Taiwan Biobank provide insights into the health and history of Han Chinese. Npj Genom. Med. 2021, 6, 10. [Google Scholar] [CrossRef]
- Lee, C.J.; Chen, T.H.; Lim, A.M.W.; Chang, C.C.; Sie, J.J.; Chen, P.L.; Chang, S.W.; Wu, S.J.; Hsu, C.L.; Hsieh, A.R.; et al. Phenome-wide analysis of Taiwan Biobank reveals novel glycemia-related loci and genetic risks for diabetes. Commun. Biol. 2022, 5, 1175. [Google Scholar] [CrossRef]
- Juang, J.M.J.; Lu, T.P.; Su, M.W.; Lin, C.W.; Yang, J.H.; Chu, H.W.; Chen, C.H.; Hsiao, Y.W.; Lee, C.Y.; Chiang, L.M.; et al. Rare variants discovery by extensive whole-genome sequencing of the Han Chinese population in Taiwan: Applications to cardiovascular medicine. J. Adv. Res. 2021, 30, 147–158. [Google Scholar] [CrossRef]
- Bonder, M.J.; Kurilshikov, A.; Tigchelaar, E.F.; Mujagic, Z.; Imhann, F.; Vila, A.V.; Deelen, P.; Vatanen, T.; Schirmer, M.; Smeekens, S.P.; et al. The effect of host genetics on the gut microbiome. Nat. Genet. 2016, 48, 1407–1412. [Google Scholar] [CrossRef]
- Imhann, F.; Bonder, M.J.; Vich Vila, A.; Fu, J.; Mujagic, Z.; Vork, L.; Tigchelaar, E.F.; Jankipersadsing, S.A.; Cenit, M.C.; Harmsen, H.J.M.; et al. Proton pump inhibitors affect the gut microbiome. Gut 2016, 65, 740–748. [Google Scholar] [CrossRef]
- Zhernakova, D.V.; Le, T.H.; Kurilshikov, A.; Atanasovska, B.; Bonder, M.J.; Sanna, S.; Claringbould, A.; Võsa, U.; Deelen, P.; Franke, L.; et al. Individual variations in cardiovascular-disease-related protein levels are driven by genetics and gut microbiome. Nat. Genet. 2018, 50, 1524–1532. [Google Scholar] [CrossRef]
- Nam, K.; Kim, J.; Lee, S. Genome-wide study on 72,298 individuals in Korean biobank data for 76 traits. Cell Genom. 2022, 2, 100189. [Google Scholar] [CrossRef]
- Moon, S.; Kim, Y.J.; Han, S.; Hwang, M.Y.; Shin, D.M.; Park, M.Y.; Lu, Y.; Yoon, K.; Jang, H.M.; Kim, Y.K.; et al. The Korea Biobank Array: Design and Identification of Coding Variants Associated with Blood Biochemical Traits. Sci. Rep. 2019, 9, 1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonfiglio, F.; Henström, M.; Nag, A.; Hadizadeh, F.; Zheng, T.; Cenit, M.C.; Tigchelaar, E.; Williams, F.; Reznichenko, A.; Ek, W.E.; et al. A GWAS meta-analysis from 5 population-based cohorts implicates ion channel genes in the pathogenesis of irritable bowel syndrome. Neurogastroenterol. Motil. 2018, 30, e13358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, J.B.; Thorolfsdottir, R.B.; Fritsche, L.G.; Zhou, W.; Skov, M.W.; Graham, S.E.; Herron, T.J.; McCarthy, S.; Schmidt, E.M.; Sveinbjornsson, G.; et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat. Genet. 2018, 50, 1234–1239. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.B.; Rom, O.; Surakka, I.; Graham, S.E.; Zhou, W.; Roychowdhury, T.; Fritsche, L.G.; Gagliano Taliun, S.A.; Sidore, C.; Liu, Y.; et al. Loss-of-function genomic variants highlight potential therapeutic targets for cardiovascular disease. Nat. Commun. 2020, 11, 6417. [Google Scholar] [CrossRef]
- Surakka, I.; Fritsche, L.G.; Zhou, W.; Backman, J.; Kosmicki, J.A.; Lu, H.; Brumpton, B.; Nielsen, J.B.; Gabrielsen, M.E.; Skogholt, A.H.; et al. MEPE loss-of-function variant associates with decreased bone mineral density and increased fracture risk. Nat. Commun. 2020, 11, 4093. [Google Scholar] [CrossRef]
- Lona-Durazo, F.; Mendes, M.; Thakur, R.; Funderburk, K.; Zhang, T.; Kovacs, M.A.; Choi, J.; Brown, K.M.; Parra, E.J. A large Canadian cohort provides insights into the genetic architecture of human hair colour. Commun. Biol. 2021, 4, 1253. [Google Scholar] [CrossRef]
- Joseph, C.B.; Mariniello, M.; Yoshifuji, A.; Schiano, G.; Lake, J.; Marten, J.; Richmond, A.; Huffman, J.E.; Campbell, A.; Harris, S.E.; et al. Meta-GWAS Reveals Novel Genetic Variants Associated with Urinary Excretion of Uromodulin. J. Am. Soc. Nephrol. 2022, 33, 511–529. [Google Scholar] [CrossRef]
- Zheng, N.S.; Stone, C.A.; Jiang, L.; Shaffer, C.M.; Kerchberger, V.E.; Chung, C.P.; Feng, Q.; Cox, N.J.; Stein, C.M.; Roden, D.M.; et al. High-throughput framework for genetic analyses of adverse drug reactions using electronic health records. PLoS Genet. 2021, 17, e1009593. [Google Scholar] [CrossRef]
- Goldstein, J.A.; Weinstock, J.S.; Bastarache, L.A.; Larach, D.B.; Fritsche, L.G.; Schmidt, E.M.; Brummett, C.M.; Kheterpal, S.; Abecasis, G.R.; Denny, J.C.; et al. LabWAS: Novel findings and study design recommendations from a meta-analysis of clinical labs in two independent biobanks. PLoS Genet. 2020, 16, e1009077. [Google Scholar] [CrossRef]
- Krebs, K.; Bovijn, J.; Zheng, N.; Lepamets, M.; Censin, J.C.; Jürgenson, T.; Särg, D.; Abner, E.; Laisk, T.; Luo, Y.; et al. Genome-wide Study Identifies Association between HLA-B*55:01 and Self-Reported Penicillin Allergy. Am. J. Hum. Genet. 2020, 107, 612–621. [Google Scholar] [CrossRef]
- Park, J.; Lucas, A.M.; Zhang, X.; Chaudhary, K.; Cho, J.H.; Nadkarni, G.; Dobbyn, A.; Chittoor, G.; Josyula, N.S.; Katz, N.; et al. Exome-wide evaluation of rare coding variants using electronic health records identifies new gene–phenotype associations. Nat. Med. 2021, 27, 66–72. [Google Scholar] [CrossRef]
- Akbari, P.; Sosina, O.A.; Bovijn, J.; Landheer, K.; Nielsen, J.B.; Kim, M.; Aykul, S.; De, T.; Haas, M.E.; Hindy, G.; et al. Multiancestry exome sequencing reveals INHBE mutations associated with favorable fat distribution and protection from diabetes. Nat. Commun. 2022, 13, 4844. [Google Scholar] [CrossRef]
- Ollier, W.; Sprosen, T.; Peakman, T. UK Biobank: From concept to reality. Pharmacogenomics 2005, 6, 639–646. [Google Scholar] [CrossRef]
- Rusk, N. The UK Biobank. Nat. Methods 2018, 15, 1001. [Google Scholar] [CrossRef]
- Sudlow, C.; Gallacher, J.; Allen, N.; Beral, V.; Burton, P.; Danesh, J.; Downey, P.; Elliott, P.; Green, J.; Landray, M.; et al. UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age. PLoS Med. 2015, 12, e1001779. [Google Scholar] [CrossRef] [Green Version]
- Nagai, A.; Hirata, M.; Kamatani, Y.; Muto, K.; Matsuda, K.; Kiyohara, Y.; Ninomiya, T.; Tamakoshi, A.; Yamagata, Z.; Mushiroda, T.; et al. Overview of the BioBank Japan Project: Study design and profile. J. Epidemiol. 2017, 27, S2–S8. [Google Scholar] [CrossRef]
- Locke, A.E.; Steinberg, K.M.; Chiang, C.W.K.; Service, S.K.; Havulinna, A.S.; Stell, L.; Pirinen, M.; Abel, H.J.; Chiang, C.C.; Fulton, R.S.; et al. Exome sequencing of Finnish isolates enhances rare-variant association power. Nature 2019, 572, 323–328. [Google Scholar] [CrossRef]
- Leitsalu, L.; Alavere, H.; Tammesoo, M.L.; Leego, E.; Metspalu, A. Linking a Population Biobank with National Health Registries—The Estonian Experience. J. Pers. Med. 2015, 5, 96–106. [Google Scholar] [CrossRef]
- Leitsalu, L.; Haller, T.; Esko, T.; Tammesoo, M.L.; Alavere, H.; Snieder, H.; Perola, M.; Ng, P.C.; Mägi, R.; Milani, L.; et al. Cohort Profile: Estonian Biobank of the Estonian Genome Center, University of Tartu. Int. J. Epidemiol. 2015, 44, 1137–1147. [Google Scholar] [CrossRef] [Green Version]
- Pormeister, K. Regulatory Environment for Biobanking in Estonia. In GDPR and Biobanking: Individual Rights, Public Interest and Research Regulation across Europe; Slokenberga, S., Tzortzatou, O., Reichel, J., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 227–242. [Google Scholar] [CrossRef]
- Wang, L.; Kong, L.; Wu, F.; Bai, Y.; Burton, R. Preventing chronic diseases in China. Lancet 2005, 366, 1821–1824. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lee, L.; Chen, J.; Collins, R.; Wu, F.; Guo, Y.; Linksted, P.; Peto, R. Cohort Profile: The Kadoorie Study of Chronic Disease in China (KSCDC). Int. J. Epidemiol. 2005, 34, 1243–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, R.G.; Millwood, I.Y.; Lin, K.; Valle, D.S.; McDonnell, P.; Hacker, A.; Avery, D.; Cai, N.; Kretzschmar, W.W.; Ansari, M.A.; et al. Genotyping and population structure of the China Kadoorie Biobank. medRxiv 2022. [Google Scholar] [CrossRef]
- Minegishi, N.; Nishijima, I.; Nobukuni, T.; Kudo, H.; Ishida, N.; Terakawa, T.; Kumada, K.; Yamashita, R.; Katsuoka, F.; Ogishima, S.; et al. Biobank Establishment and Sample Management in the Tohoku Medical Megabank Project. Tohoku J. Exp. Med. 2019, 248, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuriyama, S.; Yaegashi, N.; Nagami, F.; Arai, T.; Kawaguchi, Y.; Osumi, N.; Sakaida, M.; Suzuki, Y.; Nakayama, K.; Hashizume, H.; et al. The Tohoku Medical Megabank Project: Design and Mission. J. Epidemiol. 2016, 26, 493–511. [Google Scholar] [CrossRef] [Green Version]
- Hozawa, A.; Tanno, K.; Nakaya, N.; Nakamura, T.; Tsuchiya, N.; Hirata, T.; Narita, A.; Kogure, M.; Nochioka, K.; Sasaki, R.; et al. Study Profile of the Tohoku Medical Megabank Community-Based Cohort Study. J. Epidemiol. 2021, 31, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Turpeinen, M.; Zanger, U.M. Cytochrome P450 2B6: Function, genetics, and clinical relevance. Drug Metab. Drug Interact. 2012, 27, 185–197. [Google Scholar] [CrossRef]
- Feng, Y.C.A.; Chen, C.Y.; Chen, T.T.; Kuo, P.H.; Hsu, Y.H.; Yang, H.I.; Chen, W.J.; Su, M.W.; Chu, H.W.; Shen, C.Y.; et al. Taiwan Biobank: A rich biomedical research database of the Taiwanese population. Cell Genom. 2022, 2, 100197. [Google Scholar] [CrossRef]
- Lee, Y.C.; Chung, C.P.; Chang, M.H.; Wang, S.J.; Liao, Y.C. NOTCH3 cysteine-altering variant is an important risk factor for stroke in the Taiwanese population. Neurology 2019, 94, e87–e96. [Google Scholar] [CrossRef]
- Lin, J.C.; Hsiao, W.W.W.; Fan, C.T. Managing “incidental findings” in biobank research: Recommendations of the Taiwan biobank. Comput. Struct. Biotechnol. J. 2019, 17, 1135–1142. [Google Scholar] [CrossRef]
- Scholtens, S.; Smidt, N.; Swertz, M.A.; Bakker, S.J.; Dotinga, A.; Vonk, J.M.; van Dijk, F.; van Zon, S.K.; Wijmenga, C.; Wolffenbuttel, B.H.; et al. Cohort Profile: LifeLines, a three-generation cohort study and biobank. Int. J. Epidemiol. 2015, 44, 1172–1180. [Google Scholar] [CrossRef] [Green Version]
- Klijs, B.; Scholtens, S.; Mandemakers, J.J.; Snieder, H.; Stolk, R.P.; Smidt, N. Representativeness of the LifeLines Cohort Study. PLoS ONE 2015, 10, e0137203. [Google Scholar] [CrossRef]
- Zika, E.; Paci, D.; Schulte in den Bäumen, T.; Braun, A. Biobanks in Europe: Prospects for Harmonisation and Networking; Joint Research Centre: Ispra, Italy, 2010; p. 170. [Google Scholar] [CrossRef]
- Magnusson, P.K.E.; Almqvist, C.; Rahman, I.; Ganna, A.; Viktorin, A.; Walum, H.; Halldner, L.; Lundström, S.; Ullén, F.; Långström, N.; et al. The Swedish Twin Registry: Establishment of a Biobank and Other Recent Developments. Twin Res. Hum. Genet. 2013, 16, 317–329. [Google Scholar] [CrossRef]
- Polderman, T.J.C.; Benyamin, B.; de Leeuw, C.A.; Sullivan, P.F.; van Bochoven, A.; Visscher, P.M.; Posthuma, D. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat. Genet. 2015, 47, 702–709. [Google Scholar] [CrossRef] [Green Version]
- Beyder, A.; Mazzone, A.; Strege, P.R.; Tester, D.J.; Saito, Y.A.; Bernard, C.E.; Enders, F.T.; Ek, W.E.; Schmidt, P.T.; Dlugosz, A.; et al. Loss-of-Function of the Voltage-Gated Sodium Channel NaV1.5 (Channelopathies) in Patients With Irritable Bowel Syndrome. Gastroenterology 2014, 146, 1659–1668. [Google Scholar] [CrossRef] [Green Version]
- Hong, K.W.; Lim, J.E.; Kim, J.W.; Tabara, Y.; Ueshima, H.; Miki, T.; Matsuda, F.; Cho, Y.S.; Kim, Y.; Oh, B. Identification of three novel genetic variations associated with electrocardiographic traits (QRS duration and PR interval) in East Asians. Hum. Mol. Genet. 2014, 23, 6659–6667. Available online: http://xxx.lanl.gov/abs/https://academic.oup.com/hmg/article-pdf/23/24/6659/13907847/ddu374.pdf (accessed on 27 November 2022). [CrossRef] [Green Version]
- Cho, Y.S.; Chen, C.H.; Hu, C.; Long, J.; Hee Ong, R.T.; Sim, X.; Takeuchi, F.; Wu, Y.; Go, M.J.; Yamauchi, T.; et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat. Genet. 2012, 44, 67–72. [Google Scholar] [CrossRef]
- Kim, Y.; Han, B.G.; the KoGES group. Cohort Profile: The Korean Genome and Epidemiology Study (KoGES) Consortium. Int. J. Epidemiol. 2016, 46, e20. Available online: http://xxx.lanl.gov/abs/https://academic.oup.com/ije/article-pdf/46/2/e20/24171760/dyv316.pdf (accessed on 27 November 2022). [CrossRef]
- Krokstad, S.; Langhammer, A.; Hveem, K.; Holmen, T.; Midthjell, K.; Stene, T.; Bratberg, G.; Heggland, J.; Holmen, J. Cohort Profile: The HUNT Study, Norway. Int. J. Epidemiol. 2013, 42, 968–977. [Google Scholar] [CrossRef] [Green Version]
- Åsvold, B.O.; Langhammer, A.; Rehn, T.A.; Kjelvik, G.; Grøntvedt, T.V.; Sørgjerd, E.P.; Fenstad, J.S.; Heggland, J.; Holmen, O.; Stuifbergen, M.C.; et al. Cohort Profile Update: The HUNT Study, Norway. Int. J. Epidemiol. 2022. [Google Scholar] [CrossRef]
- Huppertz, B.; Bayer, M.; Macheiner, T.; Sargsyan, K. Biobank Graz: The Hub for Innovative Biomedical Research. Open J. Bioresour. 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Chen, B.; Wang, R.; Zhu, M.; Shao, Y.; Wang, N.; Liu, X.; Zhang, T.; Jiang, F.; Wang, W.; et al. Cohort profile: Protocol and baseline survey for the Shanghai Suburban Adult Cohort and Biobank (SSACB) study. BMJ Open 2020, 10, e035430. [Google Scholar] [CrossRef] [PubMed]
- Sankar, P.L.; Parker, L.S. The Precision Medicine Initiative’s All of Us Research Program: An agenda for research on its ethical, legal, and social issues. Genet. Med. 2017, 19, 743–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anisimov, S.V.; Meshkov, A.N.; Glotov, A.S.; Borisova, A.L.; Balanovsky, O.P.; Belyaev, V.E.; Granstrem, O.K.; Grivtsova, L.Y.; Efimenko, A.Y.; Pokrovskaya, M.S.; et al. National Association of Biobanks and Biobanking Specialists: New Community for Promoting Biobanking Ideas and Projects in Russia. Biopreservation Biobanking 2021, 19, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Barbitoff, Y.; Serebryakova, E.; Nasykhova, Y.; Predeus, A.; Polev, D.; Shuvalova, A.; Vasiliev, E.; Urazov, S.; Sarana, A.; Scherbak, S.; et al. Identification of Novel Candidate Markers of Type 2 Diabetes and Obesity in Russia by Exome Sequencing with a Limited Sample Size. Genes 2018, 9, 415. [Google Scholar] [CrossRef] [Green Version]
- Vaught, J.; Kelly, A.; Hewitt, R. A Review of International Biobanks and Networks: Success Factors and Key Benchmarks. Biopreservation Biobanking 2009, 7, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Mora, M.; Angelini, C.; Bignami, F.; Bodin, A.M.; Crimi, M.; Di Donato, J.H.; Felice, A.; Jaeger, C.; Karcagi, V.; LeCam, Y.; et al. The EuroBioBank Network: 10 years of hands-on experience of collaborative, transnational biobanking for rare diseases. Eur. J. Hum. Genet. 2015, 23, 1116–1123. [Google Scholar] [CrossRef]
- Van Ommen, G.J.B.; Törnwall, O.; Bréchot, C.; Dagher, G.; Galli, J.; Hveem, K.; Landegren, U.; Luchinat, C.; Metspalu, A.; Nilsson, C.; et al. BBMRI-ERIC as a resource for pharmaceutical and life science industries: The development of biobank-based Expert Centres. Eur. J. Hum. Genet. 2015, 23, 893–900. [Google Scholar] [CrossRef]
- Holub, P.; Swertz, M.; Reihs, R.; van Enckevort, D.; Müller, H.; Litton, J.E. BBMRI-ERIC Directory: 515 Biobanks with over 60 Million Biological Samples. Biopreservation Biobanking 2016, 14, 559–562. [Google Scholar] [CrossRef]
- Tcheandjieu, C.; Xiao, K.; Tejeda, H.; Lynch, J.A.; Ruotsalainen, S.; Bellomo, T.; Palnati, M.; Judy, R.; Klarin, D.; Kember, R.L.; et al. High heritability of ascending aortic diameter and trans-ancestry prediction of thoracic aortic disease. Nat. Genet. 2022, 54, 772–782. [Google Scholar] [CrossRef]
- Mahajan, A.; Spracklen, C.N.; Zhang, W.; Ng, M.C.Y.; Petty, L.E.; Kitajima, H.; Yu, G.Z.; Rüeger, S.; Speidel, L.; Kim, Y.J.; et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet. 2022, 54, 560–572. [Google Scholar] [CrossRef]
- Sánchez-Maldonado, J.M.; Collado, R.; Cabrera-Serrano, A.J.; Ter Horst, R.; Gálvez-Montosa, F.; Robles-Fernández, I.; Arenas-Rodríguez, V.; Cano-Gutiérrez, B.; Bakker, O.; Bravo-Fernández, M.I.; et al. Type 2 Diabetes-Related Variants Influence the Risk of Developing Prostate Cancer: A Population-Based Case-Control Study and Meta-Analysis. Cancers 2022, 14, 2376. [Google Scholar] [CrossRef]
- Larsson, S.C.; Woolf, B.; Gill, D. Plasma Caffeine Levels and Risk of Alzheimer’s Disease and Parkinson’s Disease: Mendelian Randomization Study. Nutrients 2022, 14, 1697. [Google Scholar] [CrossRef]
- COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477. [Google Scholar] [CrossRef]
- Horowitz, J.E.; Kosmicki, J.A.; Damask, A.; Sharma, D.; Roberts, G.H.L.; Justice, A.E.; Banerjee, N.; Coignet, M.V.; Yadav, A.; Leader, J.B.; et al. Genome-wide analysis provides genetic evidence that ACE2 influences COVID-19 risk and yields risk scores associated with severe disease. Nat. Genet. 2022, 54, 382–392. [Google Scholar] [CrossRef]
- Butler-Laporte, G.; Povysil, G.; Kosmicki, J.; Cirulli, E.T.; Drivas, T.; Furini, S.; Saad, C.; Schmidt, A.; Olszewski, P.; Korotko, U.; et al. Exome-wide association study to identify rare variants influencing COVID-19 outcomes: Results from the Host Genetics Initiative. medRxiv 2022. [Google Scholar] [CrossRef]
- Sakaue, S.; Kanai, M.; Tanigawa, Y.; Karjalainen, J.; Kurki, M.; Koshiba, S.; Narita, A.; Konuma, T.; Yamamoto, K.; Akiyama, M.; et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 2021, 53, 1415–1424. [Google Scholar] [CrossRef]
- Sakaue, S.; Kanai, M.; Karjalainen, J.; Akiyama, M.; Kurki, M.; Matoba, N.; Takahashi, A.; Hirata, M.; Kubo, M.; Matsuda, K.; et al. Trans-biobank analysis with 676,000 individuals elucidates the association of polygenic risk scores of complex traits with human lifespan. Nat. Med. 2020, 26, 542–548. [Google Scholar] [CrossRef]
- Pirastu, N.; Cordioli, M.; Nandakumar, P.; Mignogna, G.; Abdellaoui, A.; Hollis, B.; Kanai, M.; Rajagopal, V.M.; Parolo, P.D.B.; Baya, N.; et al. Genetic analyses identify widespread sex-differential participation bias. Nat. Genet. 2021, 53, 663–671. [Google Scholar] [CrossRef]
- Carress, H.; Lawson, D.J.; Elhaik, E. Population genetic considerations for using biobanks as international resources in the pandemic era and beyond. BMC Genom. 2021, 22, 351. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.; Finucane, H.K.; Anttila, V.; Gusev, A.; Day, F.R.; Loh, P.R.; Duncan, L.; Perry, J.R.B.; Patterson, N.; Robinson, E.B.; et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015, 47, 1236–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, M.K.; van der Spek, R.A.A.; van Rheenen, W.; Morel, S.; Bourcier, R.; Hostettler, I.C.; Alg, V.S.; van Eijk, K.R.; Koido, M.; Akiyama, M.; et al. Genome-wide association study of intracranial aneurysms identifies 17 risk loci and genetic overlap with clinical risk factors. Nat. Genet. 2020, 52, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.R.; Wei, W.Q.; Roden, D.M.; Denny, J.C. Defining Phenotypes from Clinical Data to Drive Genomic Research. Annu. Rev. Biomed. Data Sci. 2018, 1, 69–92. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef] [PubMed]
- Tcheandjieu, C.; Aguirre, M.; Gustafsson, S.; Saha, P.; Potiny, P.; Haendel, M.; Ingelsson, E.; Rivas, M.A.; Priest, J.R. A phenome-wide association study of 26 mendelian genes reveals phenotypic expressivity of common and rare variants within the general population. PLoS Genet. 2020, 16, e1008802. [Google Scholar] [CrossRef]
- Cacheiro, P.; Muñoz-Fuentes, V.; Murray, S.A.; Dickinson, M.E.; Bucan, M.; Nutter, L.M.J.; Peterson, K.A.; Haselimashhadi, H.; Flenniken, A.M. Human and mouse essentiality screens as a resource for disease gene discovery. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Dong, C.; Duan, H.; Shu, Q.; Li, H. RDmap: A map for exploring rare diseases. Orphanet J. Rare Dis. 2021, 16, 1–11. [Google Scholar] [CrossRef]
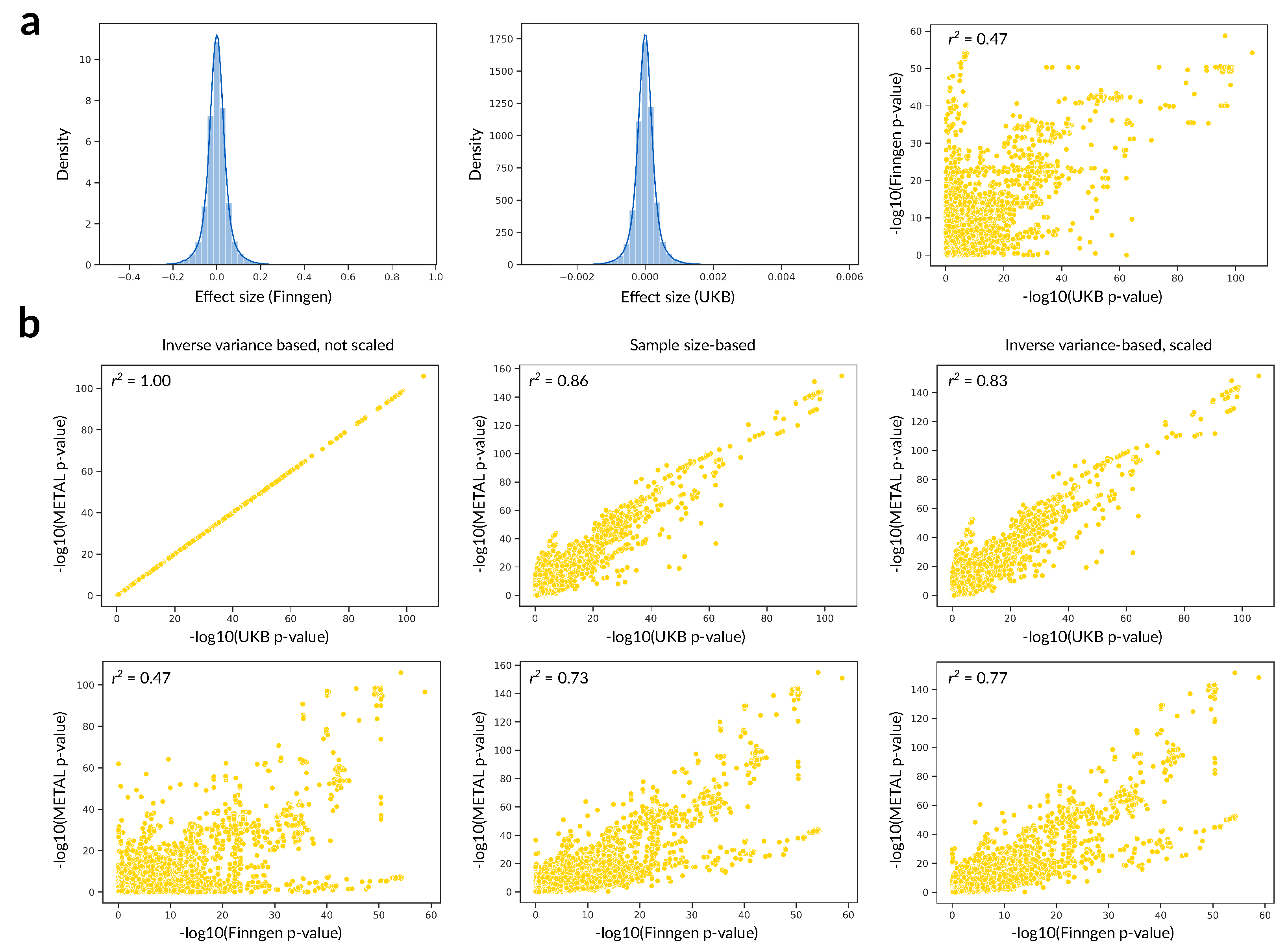
- Willer, C.J.; Li, Y.; Abecasis, G.R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010, 26, 2190–2191. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, S.; Guadagni, F.; Moore, H.; Ashton, G.; Barnes, M.; Benson, E.; Clements, J.; Koppandi, I.; Coppola, D.; Demiroglu, S.Y.; et al. Standard Preanalytical Coding for Biospecimens: Review and Implementation of the Sample PREanalytical Code (SPREC). Biopreservation Biobanking 2012, 10, 366–374. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Biobank | Location | Number of Participants a | Cohort | Biosamples | Sample Availability | Omics Data | Example Studies |
|---|---|---|---|---|---|---|---|
| UK Biobank | UK | 500,000 | closed, population aged 40–69 | blood, urine, saliva | yes | genotyping array, WGS, WES, metabolomics, telomere length | Bycroft et al., 2018 [37]; Wells et al., 2019 [38]; Watanabe et al., 2019 [26]; van Hout et al., 2020 [39]; Shikov et al., 2020 [27]; de Vincentis et al., 2022 [40]; Halldorsson et al., 2022 [15] |
| BioBank Japan | Japan | 267,307 | closed, patient-based | serum, DNA, tumor tissues | yes | genotyping array, WGS, metabolome | Ishigaki et al., 2020 [41]; Matoba et al., 2020 [42] |
| FinnGen | Finland | 538,600 | open, general population, 15 disease-specific cohorts | depends on sample collector b | depends on sample collector b | genotyping array | Desch et al., 2020 [43]; Kurki et al., 2022 [44]; Sun et al., 2022 [45] |
| Estonian biobank | Estonia | 200,000 | open, adult population | whole blood and fractions, DNA, RNA | yes | WGS, WES, genotyping array, metabolomics (NMR), RNA seq., genome-wide methylation arrays, genome-wide gene expression array | Alver et al., 2019 [29]; Reisberg et al., 2019 [46] |
| China Kadoorie Biobank | China | 512,891 | closed, residents of 5 urban and 5 rural provinces aged 30–79 | blood | no | genotyping array, WGS | Spracklen et al., 2020 [47]; Giannakopoulou et al., 2020 [48]; Zhu et al., 2021 [49]; Li et al., 2022 [50] |
| Tohoku Medical Megabank Project | Japan | 157,000 | closed, adult residents of Miyagi and Iwate Prefecture, three-generation cohort | blood fractions, urine, saliva, breast milk, dental plaque, stimulated T-cells, and EBV-transformed B-cells | yes | WGS, genotyping array, metabolomics (NMR, LC-MS), genome-wide methylation arrays | Watanabe et al., 2018 [51]; Tadaka et al., 2019 [52]; Tadaka et al., 2021 [53]; Kawame et al., 2022 [28]; Ohneda et al., 2022 [54]; Park, 2022 [55] |
| Taiwan Biobank | Taiwan | 181,635 | open, population aged 20–70, patient-based | blood, urine, saliva | yes | genotyping array, WGS, DNA methylation, HLA typing, metabolomics | Weiet al., 2021 [56]; Lee at al., 2022 [57]; Juang et al., 2021 [58] |
| LifeLines Cohort Study | Netherlands | 167,000 | closed, residents of the northern part of country | blood, urine, saliva, scalp hair | yes | genotyping array, WGS, microbiome data | Bonder et al., 2016 [59]; Imhann et al., 2016 [60]; Zhernakova et al., 2018 [61] |
| National Biobank of Korea | South Korea | 1,051,787 | population-based, patient-based | blood fractions, urine, saliva, DNA, tissue | yes | genotyping array | Namet al., 2022 [62]; Moon et al., 2019 [63] |
| Karolinska Biobank | Sweden | 700,000 | collection-specific | whole blood and fractions, urine, saliva, DNA | depends on collection | genotyping array | Bonfiglio et al., 2018 [64] |
| HUNT Biobank | Norway | 120,000 | open, adolescent and adult residents of Trøndelag | whole blood, plasma, serum, urine, saliva, feces, DNA | yes | genotyping array | Nielsen et al., 2018 [65]; Nielsen et al., 2020 [66]; Surakka et al., 2020 [67] |
| Canadian Partnership for Tomorrow’s Health | Canada | 331,359 | open, residents of 9 provinces aged 30–74 | whole blood and fractions, urine, saliva, dry blood spots, nail fragments | yes | genotyping array | Lona-Durazo et al., 2021 [68]; Joseph et al., 2022 [69] |
| All of Us Research Program | USA | 348,000 | open, adult minority population | whole blood, urine, saliva | no | WGS, genotyping array | n.a. |
| BioVU | USA | 275,000 | open, pediatric and adult patient-based | DNA | yes | genotyping array | Zhenget al., 2021 [70]; Goldstein et al., 2020 [71]; Krebs et al., 2020 [72] |
| Penn Medicine BioBank | USA | 52,853 | open, adult patient-based | blood, tissue | yes | genotyping array, WES | Parket al., 2021 [73]; Akbari et al., 2022 [74] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazareva, T.E.; Barbitoff, Y.A.; Changalidis, A.I.; Tkachenko, A.A.; Maksiutenko, E.M.; Nasykhova, Y.A.; Glotov, A.S. Biobanking as a Tool for Genomic Research: From Allele Frequencies to Cross-Ancestry Association Studies. J. Pers. Med. 2022, 12, 2040. https://doi.org/10.3390/jpm12122040
Lazareva TE, Barbitoff YA, Changalidis AI, Tkachenko AA, Maksiutenko EM, Nasykhova YA, Glotov AS. Biobanking as a Tool for Genomic Research: From Allele Frequencies to Cross-Ancestry Association Studies. Journal of Personalized Medicine. 2022; 12(12):2040. https://doi.org/10.3390/jpm12122040
Chicago/Turabian StyleLazareva, Tatyana E., Yury A. Barbitoff, Anton I. Changalidis, Alexander A. Tkachenko, Evgeniia M. Maksiutenko, Yulia A. Nasykhova, and Andrey S. Glotov. 2022. "Biobanking as a Tool for Genomic Research: From Allele Frequencies to Cross-Ancestry Association Studies" Journal of Personalized Medicine 12, no. 12: 2040. https://doi.org/10.3390/jpm12122040