Prevention of Post-Transplant Diabetes Mellitus: Towards a Personalized Approach


Abstract
:1. Introduction
2. Rationale for Prevention
2.1. Identification of at-Risk Patients
2.1.1. Metabolic Evaluation
2.1.2. Genetics
2.1.3. Integrative Score
2.2. Pathogenesis
2.3. Diagnostic Tools
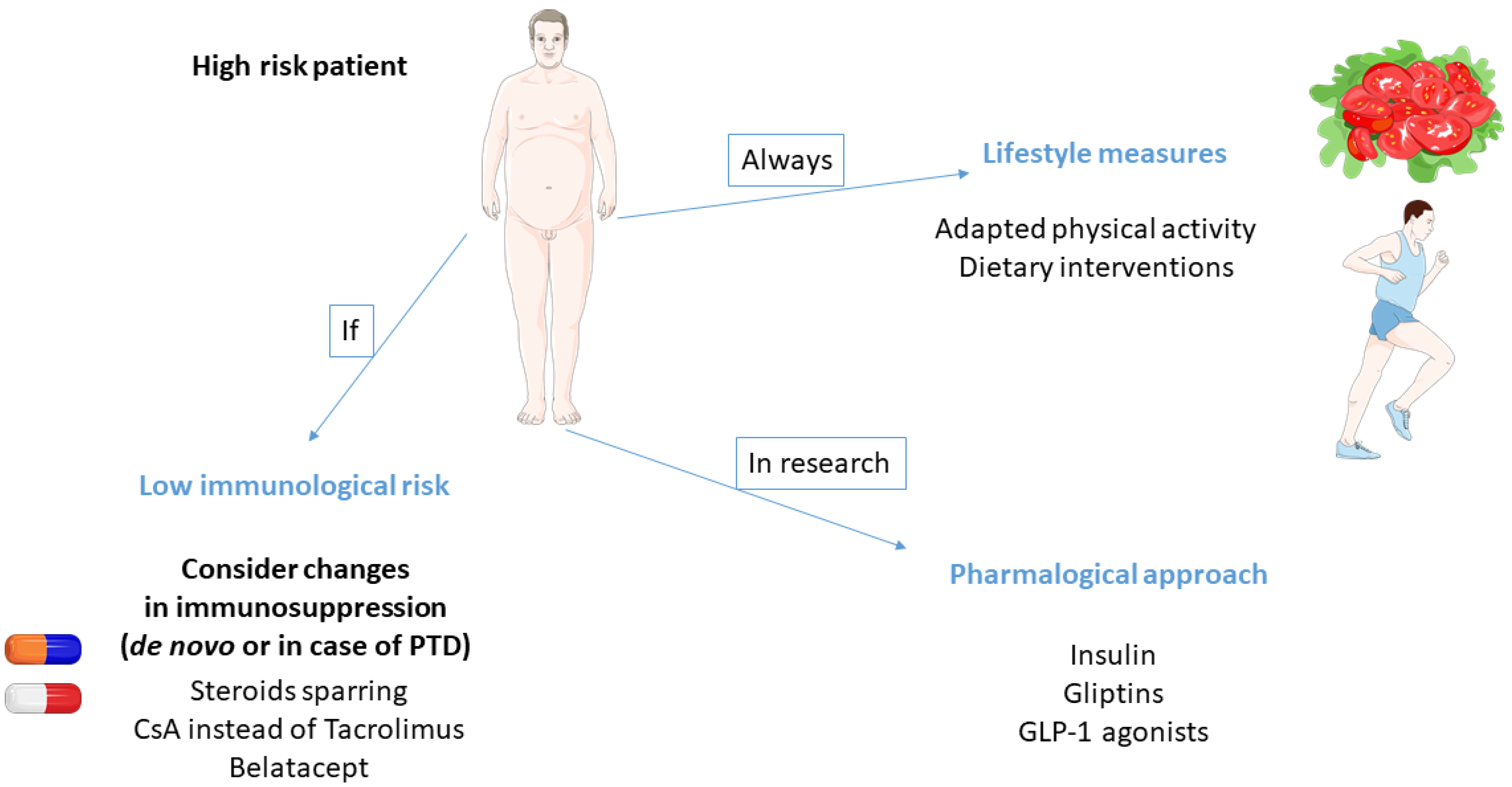
3. Prevention
3.1. Dietary Interventions
3.2. Bariatric Surgery
3.3. Physical Activity
3.4. Microbiota
3.5. Pharmacological Approach
3.5.1. Glucotoxicity and Beta-Cell Protection
3.5.2. Pharmacological Interventions to Prevent PTD
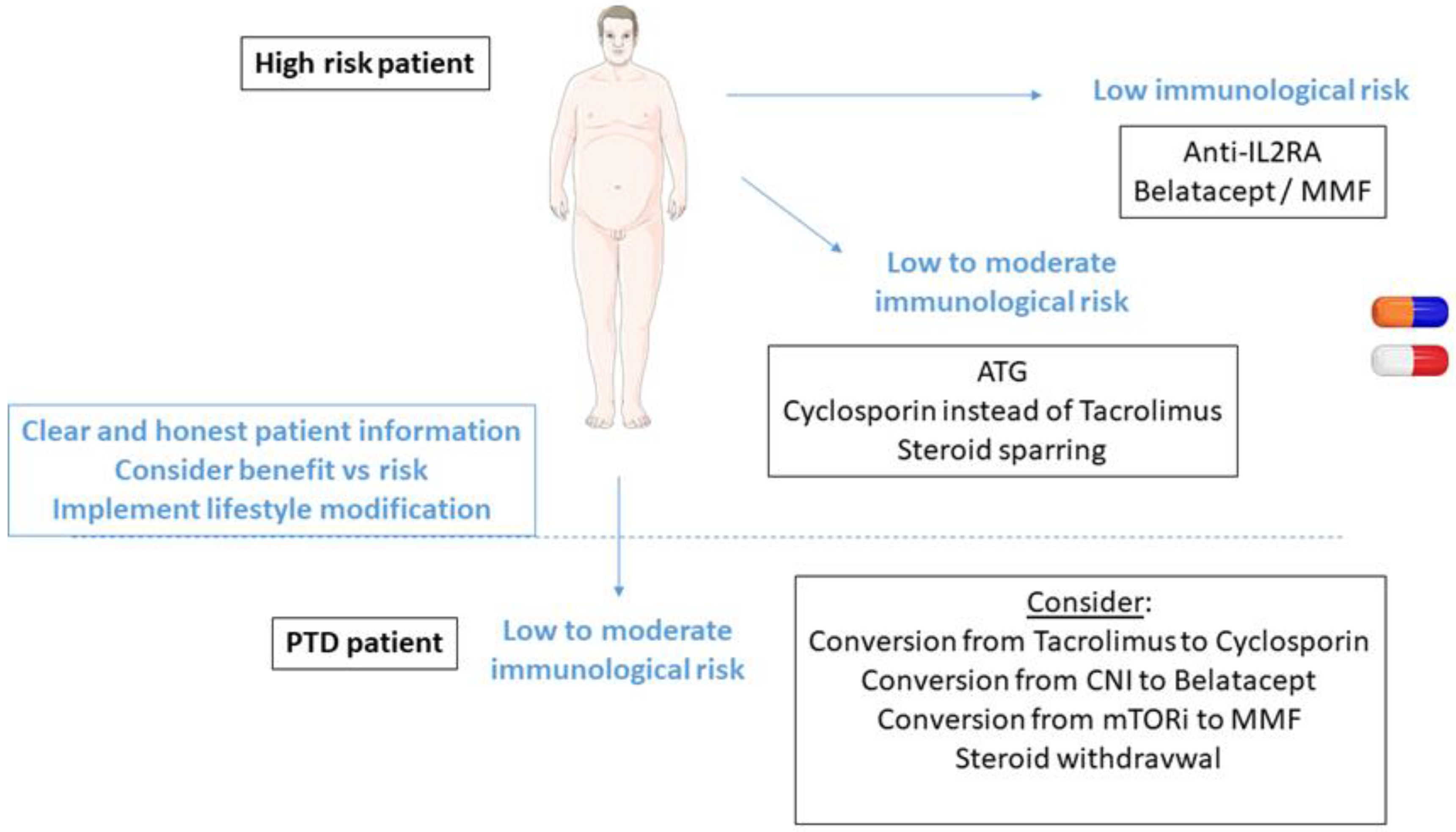
3.6. Individualization of Immunosuppression
3.6.1. Steroid-Sparing Protocols
3.6.2. Cyclosporin vs. Tacrolimus
3.6.3. mTOR Inhibitors
3.6.4. Belatacept
3.6.5. Global Immunosuppressive Strategies in High-Risk Patients
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Davidson, J.; Wilkinson, A.; Dantal, J.; Dotta, F.; Haller, H.; Hernandez, D.; Kasiske, B.L.; Kiberd, B.; Krentz, A.; Legendre, C.; et al. New-onset diabetes after transplantation: 2003 international consensus guidelines. Proceedings of an international expert panel meeting, Barcelona, Spain, 19 February 2003. Transplantation 2003, 75 (Suppl. 10), SS3–SS24. [Google Scholar] [CrossRef] [PubMed]
- Sharif, A.; Cohney, S. Post-transplant diabetes-state of the art. Lancet Diabetes Endocrinol. 2016, 37, 37–61. [Google Scholar]
- Yates, C.J.; Fourlanos, S.; Hjelmesaeth, J.; Colman, P.G.; Cohney, S.J. New-onset diabetes after kidney transplantation-changes and challenges. Am. J. Transplant. 2011, 12, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Ducloux, D.; Kazory, A.; Chalopin, J.-M. Posttransplant diabetes mellitus and atherosclerotic events in renal transplant recipients: A prospective study. Transplantation 2005, 79, 438–443. [Google Scholar] [CrossRef] [Green Version]
- Conte, C.; Secchi, A. Post-transplantation diabetes in kidney transplant recipients: An update on management and prevention. Acta Diabetol. 2018, 55, 763–779. [Google Scholar] [CrossRef]
- Eide, I.A.; Halden, T.A.S.; Hartmann, A.; Åsberg, A.; Dahle, D.O.; Reisaeter, A.V.; Jenssen, T. Mortality risk in post-transplantation diabetes mellitus based on glucose and HbA1c diagnostic criteria. Transpl. Int. 2016, 29, 568–578. [Google Scholar] [CrossRef] [Green Version]
- Eide, I.A.; Halden, T.A.S.; Hartmann, A.; Dahle, D.O.; Åsberg, A.; Jenssen, T. Associations between posttransplantation diabetes mellitus and renal graft survival. Transplantation 2017, 101, 1282–1289. [Google Scholar] [CrossRef]
- Cole, E.H.; Johnston, O.; Rose, C.L.; Gill, J.S. Impact of acute rejection and new-onset diabetes on long-term transplant graft and patient survival. Clin. J. Am. Soc. Nephrol. 2008, 3, 814–821. [Google Scholar] [CrossRef] [Green Version]
- Kuo, H.T.; Sampaio, M.S.; Vincenti, F.; Bunnapradist, S. Associations of pretransplant diabetes mellitus, new-onset diabetes after transplant, and acute rejection with transplant outcomes, an analysis of the Organ Procurement and Transplant Net-work/United Network for Organ Sharing (OPTN/UNOS) database. Am. J. Kidney Dis. 2010, 56, 1127–1139. [Google Scholar] [CrossRef]
- Valderhaug, T.G.; Hjelmesæth, J.; Jenssen, T.; Røislien, J.; Leivestad, T.; Hartmann, A. Early posttransplantation hyperglycemia in kidney transplant recipients is associated with overall long-term graft losses. Transplantation 2012, 94, 714–720. [Google Scholar] [CrossRef]
- Courivaud, C.; Ladriere, M.; Toupance, O.; Caillard, S.; De Ligny, B.H.; Ryckelynck, J.-P.; Moulin, B.; Rieu, P.; Frimat, L.; Chalopin, J.-M.; et al. Impact of pre-transplant dialysis modality on post-transplant diabetes mellitus after kidney transplantation. Clin. Transplant. 2010, 25, 794–799. [Google Scholar] [CrossRef] [PubMed]
- von Düring, M.E.; Jenssen, T.; Bollerslev, J.; Åsberg, A.; Godang, K.; Eide, I.A.; Dahle, D.O.; Hartmann, A. Visceral fat is better related to impaired glucose metabolism than body mass index after kidney transplantation. Transplant. Int. 2015, 28, 1162–1171. [Google Scholar] [CrossRef]
- Kim, J.E.; Park, S.J.; Kim, Y.C.; Min, S.-I.; Ha, J.; Kim, Y.S.; Yoon, S.H.; Han, S.S. Deep learning-based quantification of visceral fat volumes predicts posttransplant diabetes mellitus in kidney transplant recipients. Front. Med. 2021, 8, 632097. [Google Scholar] [CrossRef] [PubMed]
- Gomes, V.; Ferreira, F.; Guerra, J.; Bugalho, M.J. New-onset diabetes after kidney transplantation: Incidence and associated factors. World J. Diabetes 2018, 9, 132–137. [Google Scholar] [CrossRef]
- Hap, K.; Madziarska, K.; Zmonarski, S.; Kamińska, D.; Magott-Procelewska, M.; Banasik, M.; Jędrzejak, K.; Madziarski, M.; Hap, W.; Klinger, M.; et al. Pretransplantation oral glucose tolerance test can prevent posttransplant diabetes mellitus after renal transplantation: Preliminary study. Transplant. Proc. 2018, 50, 1776–1780. [Google Scholar] [CrossRef] [PubMed]
- Tokodai, K.; Amada, N.; Haga, I.; Takayama, T.; Nakamura, A. The 5-time point oral glucose tolerance test as a predictor of new-onset diabetes after kidney transplantation. Diabetes Res. Clin. Pract. 2014, 103, 298–303. [Google Scholar] [CrossRef]
- Iida, S.; Ishida, H.; Tokumoto, T.; Omoto, K.; Shirakawa, H.; Shimizu, T.; Amano, H.; Setoguchi, K.; Nozaki, T.; Toki, D.; et al. New-onset diabetes after transplantation in tacrolimus-treated, living kidney transplantation: Long-term impact and utility of the pre-transplant OGTT. Int. Urol. Nephrol. 2010, 42, 935–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh Prasad, G.V.; Huang, M.; Bandukwala, F.; Nash, M.M.; Rapi, L.; Montada-Atin, T.; Meliton, G.; Zaltzman, J.S. Pre-transplantation glucose testing for predicting new- onset diabetes mellitus after renal transplantation. Clin. Nephrol. 2009, 71, 140–146. [Google Scholar]
- McCaughan, J.A.; McKnight, A.J.; Maxwell, A.P. Genetics of New-Onset Diabetes after Transplantation. J. Am. Soc. Nephrol. 2013, 25, 1037–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamoulid, J.; Courivaud, C.; Deschamps, M.; Mercier, P.; Ferrand, C.; Penfornis, A.; Tiberghien, P.; Chalopin, J.-M.; Saas, P.; Ducloux, D. IL-6 promoter polymorphism—174 is associated with new-onset diabetes after transplantation. J. Am. Soc. Nephrol. 2006, 17, 2333–2340. [Google Scholar] [CrossRef]
- Quaglia, M.; Terrazzino, S.; Musetti, C.; Cargnin, S.; Merlotti, G.; Cena, T.; Stratta, P.; Genazzani, A. The role of TCF7L2 rs7903146 in diabetes after kidney transplant, results from a single-center cohort and meta-analysis of the literature. Transplantation 2016, 100, 1750–1758. [Google Scholar] [CrossRef]
- Dabrowska-Zamojcin, E.; Romanowski, M.; Dziedziejko, V.; Maciejewska-Karlowska, A.; Sawczuk, M.; Safranow, K.; Domanski, L.; Pawlik, A. CCL2 gene polymorphism is associated with post-transplant diabetes mellitus. Int. Immunopharmacol. 2016, 32, 62–65. [Google Scholar] [CrossRef]
- Romanowski, M.; Dziedziejko, V.; Maciejewska-Karlowska, A.; Sawczuk, M.; Safranow, K.; Domanski, L.; Pawlik, A. Adiponectin and leptin gene polymorphisms in patients with.post-transplant diabetes mellitus. Pharmacogenomics 2015, 16, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Lee Sr Moon, J.Y.; Lee, S.H.; Ihm, C.G.; Lee, T.W.; Kim, S.K.; Chung, J.H.; Kang, S.W.; Kim, T.H.; Park, S.J.; Kim, Y.H.; et al. Angioten-sinogen polymorphisms and post-transplantation diabetes mellitus in Korean renal transplant subjects. Kidney. Blood Press Res. 2013, 37, 95–102. [Google Scholar]
- Chakkera, H.A.; Weil, E.J.; Swanson, C.M.; Dueck, A.C.; Heilman, R.L.; Reddy, K.S.; Hamawi, K.; Khamash, H.; Moss, A.A.; Mulligan, D.C.; et al. Pretransplant risk score for new-onset diabetes after kidney transplantation. Diabetes Care 2011, 34, 2141–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakkera, H.A.; Chang, Y.H.; Ayub, A.; Gonwa, T.A.; Weil, E.J.; Knowler, W.C. Validation of apretransplant risk score for new-onset diabetes after kidney transplantation. Diabetes Care 2013, 36, 2881–2886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagen, M.; Hjelmesæth, J.; Jenssen, T.; Mørkrid, L.; Hartmann, A. A 6-year prospective study on new onset diabetes mellitus, insulin release and insulin sensitivity in renal transplant recipients. Nephrol. Dial. Transplant. 2003, 18, 2154–2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duijnhoven, E.M.V.; Boots, J.M.M.; Christiaans, M.H.L.; Wolffenbuttel, B.H.R.; Hooff, J.P.V. Influence of tacrolimus on glucose metab-olism before and after renal transplantation, a prospective study. J. Am. Soc. Nephrol. 2001, 12, 583–588. [Google Scholar] [CrossRef]
- Tamura, K.; Fujimura, T.; Tsutsumi, T.; Nakamura, K.; Ogawa, T.; Atumaru, C.; Hirano, Y.; Ohara, K.; Ohtsuka, K.; Shimomura, K. Transcriptional inhibition of insulin by FK506 and possible involvement of FK506 binding protein-12 in pancreatic beta-cell. Transplantation 1995, 59, 1606–1613. [Google Scholar]
- Sharif, A.; Hecking, M.; de Vries, A.P.; Porrini, E.; Hornum, M.; Rasoul-Rockenschaub, S.; Berlakovich, G.; Krebs, M.; Kautzky-Willer, A.; Schernthaner, G.; et al. Proceedings from an international consensus meeting on posttransplan-tation diabetes mellitus, recommendations and future directions. Am. J. Transplant. 2014, 14, 1992–2000. [Google Scholar] [CrossRef] [Green Version]
- Hoban, R.; Gielda, B.; Temkit, M.; Saha, C.; Book, B.K.; Baker, E.; Pescovitz, M.D. Utility of HbA1c in the Detection of Subclinical Post Renal Transplant Diabetes. Transplantation 2006, 81, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Shabir, S.; Jham, S.; Harper, L.; Ball, S.; Borrows, R.; Sharif, A. Validity of glycated haemoglobin to diagnose new onset diabetes after transplantation. Transpl. Int. 2013, 26, 315–321. [Google Scholar] [CrossRef]
- Gomes-Neto, A.W.; Osté, M.C.J.; Sotomayor, C.G.; VD Berg, E.; Geleijnse, J.M.; Gans, R.O.B.; Bakker, S.J.L.; Navis, G.J. Fruit and vegetable intake and risk of posttransplantation diabetes in renal transplant recipients. Diabetes Care 2019, 42, 1645–1652. [Google Scholar] [CrossRef]
- Osté, M.C.J.; Corpeleijn, E.; Navis, G.J.; Keyzer, C.A.; Soedamah-Muthu, S.S.; van den Berg, E.; Postmus, D.; de Borst, M.H.; Kromhout, D.; Bakker, S.J.L. Mediterranean style diet is associated with low risk of new-onset diabetes after renal transplantation. BMJ Open Diabetes Res. Care 2017, 5, e000283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kittiskulnam, P.; Johansen, K.L. The obesity paradox, A further consideration in dialysis patients. Semin. Dial. 2019, 32, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Rhee, C.M.; Ahmadi, S.-F.; Kalantar-Zadeh, K. The dual roles of obesity in chronic kidney disease, a review of the current literature. Curr. Opin. Nephrol. Hypertens. 2016, 25, 208–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conley, M.M.; McFarlane, C.M.; MacLaughlin, H.L.; Johnson, D.W.; Campbell, K.L. Interventions for weight loss in people with chronic kidney disease who are overweight or obese. Cochrane Database Syst. Rev. 2018, 3, 013119. [Google Scholar] [CrossRef] [Green Version]
- Cook, S.A.; MacLaughlin, H.; MacDougall, I.C. A structured weight management programme can achieve improved functional ability and significant weight loss in obese patients with chronic kidney disease. Nephrol. Dial. Transplant. 2007, 23, 263–268. [Google Scholar] [CrossRef]
- Kuningas, K.; Driscoll, J.; Mair, R.; Smith, H.; Dutton, M.; Day, E.; Sharif, A.A. Comparing glycaemic benefits of active versus passive lifestyle intervention in kidney allograft recipients: A randomized controlled trial. Transplantation 2020, 104, 1491–1499. [Google Scholar] [CrossRef]
- Kassam, A.F.; Mirza, A.; Kim, Y.; Hanseman, D.; Woodle, E.S.; Quillin, R.C., 3rd; Johnson, B.L.; Govil, A.; Cardi, M.; Schauer, D.P.; et al. Long-term outcomes in patients with obesity and renal disease after sleeve gastrectomy. Am. J. Transplant. 2020, 20, 422–429. [Google Scholar] [CrossRef]
- Yemini, R.; Nesher, E.; Carmeli, I.; Winkler, J.; Rahamimov, R.; Mor, E.; Keidar, A. Bariatric surgery is efficacious and improves access to transplantation for morbidly obese renal transplant candidates. Obes. Surg. 2019, 29, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Carandina, S.; Genser, L.; Bossi, M.; Montana, L.; Cortes, A.; Seman, M.; Danan, M.; Barrat, C. Laparoscopic sleeve gastrectomy in kidney transplant candidates: A case series. Obes. Surg. 2017, 27, 2613–2618. [Google Scholar] [CrossRef] [PubMed]
- Soliman, B.G.; Tariq, N.; Law, Y.Y.; Yi, S.; Nwana, N.; Bosetti, R.; Kash, B.; Moore, L.W.; Gaber, A.O.; Sherman, V. Effectiveness of bariatric surgery in increasing kidney transplant eligibility in patients with kidney failure requiring dialysis. Obes. Surg. 2021, 31, 3436–3443. [Google Scholar] [CrossRef]
- Kim, Y.; Jung, A.D.; Dhar, V.K.; Tadros, J.S.; Schauer, D.P.; Smith, E.P.; Hanseman, D.J.; Cuffy, M.C.; Alloway, R.R.; Shields, A.R.; et al. Laparoscopic sleeve gastrectomy improves renal transplant candidacy and posttransplant outcomes in morbidly obese patients. Am. J. Transplant. 2017, 18, 410–416. [Google Scholar] [CrossRef]
- Clyne, N.; Anding-Rost, K. Exercise training in chronic kidney disease—effects, expectations and adherence. Clin. Kidney J. 2021, 14, ii3–ii14. [Google Scholar] [CrossRef]
- Hoshino, J. Renal rehabilitation: Exercise intervention and nutritional support in dialysis patients. Nutrients 2021, 13, 1444. [Google Scholar] [CrossRef]
- Wilkinson, T.J.; McAdams-DeMarco, M.; Bennett, P.N.; Wilund, K. Global renal exercise network. Advances in exercise therapy in predialysis chronic kidney disease, hemodialysis, peritoneal dialysis, and kidney transplantation. Curr. Opin. Nephrol. Hypertens. 2020, 29, 471–479. [Google Scholar] [CrossRef]
- Sgrò, P.; Emerenziani, G.P.; Antinozzi, C.; Sacchetti, M.; Di Luigi, L. Exercise as a drug for glucose management and prevention in type 2 diabetes mellitus. Curr. Opin. Pharmacol. 2021, 59, 95–102. [Google Scholar] [CrossRef]
- Cárdenas Fuentes, G.; Bawaked, R.A.; Martínez González, M.Á.; Corella, D.; Subirana Cachinero, I.; Salas-Salvadó, J.; Estruch, R.; Ser-ra-Majem, L.; Ros, E.; Lapetra Peralta, J.; et al. Association of physical activity with body mass index, waist circumference and incidence of obesity in older adults. Eur. J. Public Health 2018, 28, 944–950. [Google Scholar] [CrossRef]
- Whitham, M.; Chan, M.H.; Pal, M.; Matthews, V.B.; Prelovsek, O.; Lunke, S.; El-Osta, A.; Broenneke, H.; Alber, J.; Brüning, J.C.; et al. Contraction-induced interleukin-6 gene tran-scription in skeletal muscle is regulated by c-Jun terminal kinase/activator protein-1. J. Biol. Chem. 2012, 287, 10771–10779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Canoves, P.; Scheele, C.; Pedersen, B.K.; Serrano, A.L. Interleukin-6 myokine signaling in skeletal muscle, a double-edged sword? FEBS J. 2013, 280, 4131–4148. [Google Scholar] [CrossRef]
- Starkie, R.; Ostrowski, S.R.; Jauffred, S.; Febbraio, M.; Pedersen, B.K. Exercise and IL-6 infusion inhibit endotoxin-induced TNF-a production in humans. FASEB J. 2003, 17, 884–886. [Google Scholar] [CrossRef] [PubMed]
- Hemmingsen, B.; Gimenez-Perez, G.; Mauricio, D.; Figuls, M.R.I.; Metzendorf, M.-I.; Richter, B. Diet, physical activity or both for prevention or delay of type 2 diabetes mellitus and its associated complications in people at increased risk of developing type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2017, 2017, CD003054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uusitupa, M.; Khan, T.A.; Viguiliouk, E.; Kahleova, H.; Rivellese, A.A.; Hermansen, K.; Pfeiffer, A.; Thanopoulou, A.; Salas-Salvadó, J.; Schwab, U.; et al. Prevention of Type 2 diabetes by lifestyle changes: A systematic review and meta-analysis. Nutrients 2019, 11, 2611. [Google Scholar] [CrossRef] [Green Version]
- Haw, J.S.; Galaviz, K.I.; Straus, A.N.; Kowalski, A.J.; Magee, M.J.; Weber, M.B.; Ali, M.K.; Narayan, K.M.V.; Wei, J. Long-term sustainability of diabetes prevention approaches, a systematic review and meta-analysis of randomized clinical trials. JAMA Int. Med. 2017, 177, 1808–1817. [Google Scholar] [CrossRef]
- Pan, X.-R.; Li, G.-W.; Hu, Y.-H.; Wang, J.-X.; Yang, W.-Y.; An, Z.-X.; Hu, Z.-X.; Lin, J.-; Xiao, J.-Z.; Cao, H.-B.; et al. Effects of diet and exercise in preventing niddm in people with impaired glucose tolerance: The da qing igt and diabetes study. Diabetes Care 1997, 20, 537–544. [Google Scholar] [CrossRef]
- Hellgren, M.I.; Jansson, P.-A.; Wedel, H.; Lindblad, U. A lifestyle intervention in primary care prevents deterioration of insulin resistance in patients with impaired glucose tolerance: A randomised controlled trial. Scand. J. Public Health 2016, 44, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Byambasukh, O.; Osté, M.C.J.; Gomes-Neto, A.W.; van den Berg, E.; Navis, G.; Bakker, S.J.L.; Corpeleijn, E. Physical activity and the development of post-transplant diabetes mellitus, and cardiovascular- and all-cause mortality in renal transplant recipients. J. Clin. Med. 2020, 9, 415. [Google Scholar] [CrossRef] [Green Version]
- Qiu, S.; Cai, X.; Schumann, U.; Velders, M.; Sun, Z.; Steinacker, J.M. Impact of walking on glycemic control and other cardiovascular risk factors in type 2 diabetes: A meta-analysis. PLoS ONE 2014, 9, e109767. [Google Scholar] [CrossRef] [Green Version]
- Way, K.L.; Sabag, A.; Sultana, R.N.; Baker, M.K.; Keating, S.E.; Lanting, S.; Gerofi, J.; Chuter, V.H.; Caterson, I.D.; Twigg, S.M.; et al. The effect of low-volume high-density interval training on cardiovascular health outcomes in type 2 diabetes, a randomized controlled trial. Int. J. Cardiol. 2020, 320, 148–154. [Google Scholar] [CrossRef]
- Zhang, L.; Chu, J.; Hao, W.; Zhang, J.; Li, H.; Yang, C.; Yang, J.; Chen, X.; Wang, H. Gut microbiota and type 2 diabetes mellitus: Association, mechanism, and translational applications. Mediat. Inflamm. 2021, 2021, 5110276. [Google Scholar] [CrossRef]
- Xia, F.; Wen, L.-P.; Ge, B.-C.; Li, Y.-X.; Li, F.-P.; Zhou, B.-J. Gut microbiota as a target for prevention and treatment of type 2 diabetes: Mechanisms and dietary natural products. World J. Diabetes 2021, 12, 1146–1163. [Google Scholar] [CrossRef]
- Allin, K.H.; Tremaroli, V.; Caesar, R.; Jensen, B.A.H.; Damgaard, M.T.F.; Bahl, M.I.; Licht, T.R.; Hansen, T.H.; Nielsen, T.; Dantoft, T.M.; et al. Aberrant intestinal microbiota in individuals with prediabetes. Diabetologia 2018, 61, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Tremaroli, V.; Schmidt, C.; Lundqvist, A.; Olsson, L.M.; Krämer, M.; Gummesson, A.; Perkins, R.; Bergström, G.; Bäckhed, F. The gut microbiota in prediabetes and diabetes, a population-based cross-sectional study. Cell Metab. 2020, 32, 379–390.e3. [Google Scholar] [CrossRef]
- Sanna, S.; Van Zuydam, N.R.; Mahajan, A.; Kurilshikov, A.; Vila, A.V.; Võsa, U.; Mujagic, Z.; Masclee, A.A.M.; Jonkers, D.M.A.E.; Oosting, M.; et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat. Genet. 2019, 51, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Swisa, A.; Glaser, B.; Dor, Y. Metabolic stress and compromised identity of pancreatic beta cells. Front. Genet. 2017, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Maedler, K.; Spinas, G.A.; Lehmann, R.; Sergeev, P.; Weber, M.; Fontana, A.; Kaiser, N.; Donath, M.Y. Glucose induces ¦Â cell apoptosis via upregulations of the Fas-receptor in human islets. Diabetes 2001, 50, 1683–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Meier, J.J. Linking the genetics of type 2 diabetes with low birth weight: A role for prenatal islet maldevelopment? Diabetes 2009, 58, 1255–1256. [Google Scholar] [CrossRef] [Green Version]
- Pinnick, K.; Neville, M.; Clark, A.; Fielding, B. Reversibility of metabolic and morphological changes associated with chronic exposure of pancreatic islet beta-cells to fatty acids. J. Cell Biochem. 2010, 109, 683–692. [Google Scholar] [PubMed]
- Cruzat, V.F.; Keane, K.N.; Scheinpflug, A.L.; Cordeiro, R.; Soares, M.J.; Newsholme, P. Alanyl-glutamine improves pancreatic beta-cell function following ex vivo inflammatory challenge. J. Endocrinol. 2015, 224, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Lim, E.L.; Hollingsworth, K.G.; Aribisala, B.S.; Chen, M.J.; Mathers, J.C.; Taylor, R. Reversal of type 2 diabetes: Normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 2011, 54, 2506–2514. [Google Scholar] [CrossRef] [Green Version]
- Steven, S.; Hollingsworth, K.G.; Al-Mrabeh, A.; Avery, L.; Aribisala, B.; Caslake, M.; Taylor, R. Very low-calorie diet and 6 months of weight stability in type 2 diabetes: Pathophysiological changes in responders and nonresponders. Diabetes Care 2016, 39, 808–815. [Google Scholar] [CrossRef] [Green Version]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, E.A.; Imes, S.; Wallace, C. Short-term intensive insulin therapy in newly diagnosed type 2 diabetes. Diabetes Care 2004, 27, 1028–1032. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xu, W.; Liao, Z.; Yao, B.; Chen, X.; Huang, Z.; Hu, G.; Weng, J. Induction of long-term glycemic control in newly diagnosed type 2 diabetic patients is associated with improvement of beta-cell function. Diabetes Care 2004, 27, 2597–2602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecking, M.; Haidinger, M.; Döller, D.; Werzowa, J.; Tura, A.; Zhang, J.; Tekoglu, H.; Pleiner, J.; Wrba, T.; Rasoul-Rockenschaub, S.; et al. Early basal insulin therapy decreases new-onset diabetes after renal transplantation. J. Am. Soc. Nephrol. 2012, 23, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Schwaiger, E.; Krenn, S.; Kurnikowski, A.; Bergfeld, L.; Pérez-Sáez, M.J.; Frey, A.; Topitz, D.; Bergmann, M.; Hödlmoser, S.; Bachmann, F.; et al. Early postoperative basal insulin therapy versus standard of care for the prevention of diabetes mellitus after kidney transplantation: A multicenter randomized trial. J. Am. Soc. Nephrol. 2021, 32, 2083–2098. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, P.L.; Drucker, D.J. Minireview, glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology 2004, 145, 2653–2659. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; AStoffers, D.; Habener, J.F.; Bonner-Weir, S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes 1999, 48, 2270–2276. [Google Scholar] [CrossRef]
- Tourrel, C.; Bailbe, D.; Meile, M.-J.; Kergoat, M.; Portha, B. Glucagon-like peptide-1 and exendin-4 stimulate beta-cell neogenesis in streptozotocin-treated newborn rats resulting in persistently improved glucose homeostasis at adult age. Diabetes 2001, 50, 1562–1570. [Google Scholar] [CrossRef]
- Tourrel, C.; Bailbe, D.; Lacorne, M.; Meile, M.J.; Kergoat, M.; Portha, B. Persistent improvement of type 2 diabetes in the Go-to-Kakizaki rat model by expansion of the beta-cell mass during the prediabetic period with glucagon-like peptide-1 or exendin-4. Diabetes 2002, 51, 1443–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Brubaker, P. Glucagon-like peptide-1 treatment delays the onset of diabetes in 8 week-old db/db mice. Diabetologia 2002, 45, 1263–1273. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Rao, X.; Rajagopalan, S. An emerging role of dipeptidyl peptidase 4 (DPP4) beyond glucose control: Potential implications in cardiovascular disease. Atherosclerosis 2013, 226, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Haidinger, M.; Werzowa, J.; Hecking, M.; Antlanger, M.; Stemer, G.; Pleiner, J.; Kopecky, C.; Kovarik, J.J.; Döller, D.; Pacini, G.; et al. Efficacy and safety of vildagliptin in new-onset diabetes after kidney transplantation-a randomized, double-blind, placebo-controlled trial. Am. J. Transplant. 2014, 14, 115–123. [Google Scholar] [CrossRef]
- Werzowa, J.; Hecking, M.; Haidinger, M.; Lechner, F.; Döller, D.; Pacini, G.; Stemer, G.; Pleiner, J.; Frantal, S.; Säemann, M.D. Vildag-liptin and pioglitazone in patients with impaired glucose tolerance after kidney transplantation, a randomized, place-bo-controlled clinical trial. Transplantation 2013, 95, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Gaiffe, E.; Crepin, T.; Bamoulid, J.; Courivaud, C.; Büchler, M.; Cassuto, E.; Albano, L.; Chemouny, J.M.; Choukroun, G.; Hazzan, M.; et al. PRODIG (Prevention of new onset diabetes after transplantation by a short term treatment of Vildagliptin in the early renal post-transplant period) study: Study protocol for a randomized controlled study. Trials 2019, 20, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, J.; Galeano, C.; Royuela, A.; Zamora, J. A systematic review on steroid withdrawal between 3 and 6 months after kidney transplantation. Transplantation 2010, 90, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Haller, M.C.; Royuela, A.; Nagler, E.V.; Pascual, J.; Webster, A.C. Steroid avoidance or withdrawal for kidney transplant recipi-ents. Cochrane Database Syst. Rev. 2016, 2016, CD005632. [Google Scholar]
- Thomusch, O.; Wiesener, M.; Opgenoorth, M.; Pascher, A.; Woitas, R.P.; Witzke, O.; Jaenigen, B.; Rentsch, M.; Wolters, H.; Rath, T.; et al. Rabbit-ATG or basiliximab induction for rapid steroid withdrawal after renal transplan-tation (Harmony), an open-label, multicentre, randomised controlled trial. Lancet 2016, 388, 3006–3016. [Google Scholar] [CrossRef]
- Chakkera, H.A.; Kudva, Y.; Kaplan, B. Calcineurin Inhibitors: Pharmacologic mechanisms impacting both insulin resistance and insulin secretion leading to glucose dysregulation and diabetes mellitus. Clin. Pharmacol. Ther. 2017, 101, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.C.; Woodroffe, R.C.; Taylor, R.S.; Chapman, J.R.; Craig, J.C. Tacrolimus versus cyclosporin as primary immunosuppression for kidney transplant recipients. Cochrane Database Syst. Rev. 2005, 4, CD003961. [Google Scholar] [CrossRef]
- Vincenti, F.; Friman, S.; Scheuermann, E.; Rostaing, L.; Jenssen, T.; Campistol, J.M.; Uchida, K.; Pescovitz, M.D.; Marchetti, P.; Tuncer, M.; et al. Results of an international, randomized trial comparing glucose metabolism disorders and outcome with cyclosporine versus tacrolimus. Am. J. Transplant. 2007, 7, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Heisel, O.; Heisel, R.; Balshaw, R.; Keown, P. New onset diabetes mellitus in patients receiving calcineurin inhibitors: A systematic review and meta-analysis. Am. J. Transplant. 2004, 4, 583–595. [Google Scholar] [CrossRef]
- Webster, A.C.; Woodroffe, R.C.; Taylor, R.S.; Chapman, J.R.; Craig, J.C. Tacrolimus versus ciclosporin as primary immunosuppression for kidney transplant recipients, meta-analysis and meta-regression of randomized trial data. BMJ 2005, 331, 810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, A.; Hernández, D.; Moreso, F.; Serón, D.; Burgos, M.D.; Pallardó, L.M.; Kanter, J.; Díaz Corte, C.; Rodríguez, M.; Diaz, J.M.; et al. Randomized controlled trial assessing the impact of tacrolimus versus cyclosporine on the incidence of posttransplant diabetes mellitus. Kidney Int. Rep. 2018, 3, 1304–1315. [Google Scholar] [CrossRef] [Green Version]
- Wissing, K.M.; Abramowicz, D.; Weekers, L.; Budde, K.; Rath, T.; Witzke, O.; Broeders, N.; Kianda, M.; Kuypers, D.R.J. Prospective randomized study of conversion from tacrolimus to cyclosporine A to improve glucose metabolism in patients with post-transplant diabetes mellitus after renal transplantation. Am. J. Transplant. 2018, 18, 1726–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, E.; Cao, X.; Moibi, J.A.; Greene, S.R.; Young, R.; Trucco, M.; Gao, Z.; Matschinsky, F.M.; Deng, S.; Markman, J.F.; et al. Rapamycin has a deleterious effect on min-6 cells and rat and human islets. Diabetes 2003, 52, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Shivaswamy, V.; Boerner, B.; Larsen, J. Post-transplant diabetes mellitus: Causes, treatment, and impact on outcomes. Endocr. Rev. 2016, 37, 37–61. [Google Scholar] [CrossRef] [Green Version]
- Teutonico, A.; Schena, P.F.; Di Paolo, S. Glucose metabolism in renal transplant recipients, effect of calcineurin inhibitor with-drawal and conversion to sirolimus. J. Am. Soc. Nephrol. 2005, 16, 3128–3135. [Google Scholar] [CrossRef] [Green Version]
- Johnston, O.; Rose, C.L.; Webster, A.C.; Gill, J.S. Sirolimus is associated with new-onset diabetes in kidney transplant recipients. J. Am. Soc. Nephrol. 2008, 19, 1411–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qazi, Y.; Shaffer, D.; Kaplan, B.; Kim, D.Y.; Luan, F.L.; Peddi, V.R.; Shihab, F.; Tomlanovich, S.; Yilmaz, S.; McCague, K.; et al. Efficacy and safety of everolimus plus low-dose tacrolimus versus mycophenolate mofetil plus standard-dose tacrolimus inde novorenal transplant recipients: 12-month data. Arab. Archaeol. Epigr. 2017, 17, 1358–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincenti, F.; Rostaing, L.; Grinyo, J.; Rice, K.; Steinberg, S.; Gaite, L.; Moal, M.-C.; Mondragon-Ramirez, G.A.; Kothari, J.; Polinsky, M.S.; et al. Belatacept and long-term outcomes in kidney transplantation. N. Engl. J. Med. 2016, 374, 333–343. [Google Scholar] [CrossRef]
- Vanrenterghem, Y.; Bresnahan, B.; Campistol, J.; Durrbach, A.; Grinyó, J.; Neumayer, H.H.; Lang, P.; Larsen, C.P.; Mancilla-Urrea, E.; Pestana, J.M.; et al. Belatacept-based regimens are associated with improved cardiovascular and metabolic risk factors compared with cyclosporine in kidney transplant recipients (BENEFIT and BENE-FIT-EXT studies). Transplantation 2011, 91, 976–983. [Google Scholar] [CrossRef]
- Masson, P.; Henderson, L.; Chapman, J.R.; Craig, J.C.; Webster, A.C. Belatacept for kidney transplant recipients. Cochrane Data-base Syst. Rev. 2014, 2014, CD010699. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Risk of PTD | |
|---|---|
| Steroids | +++ |
| Cyclosporin | ++ |
| Tacrolimus | +++ |
| mTORi | ++ |
| AZA/MMF | 0 |
| Belatacept | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ducloux, D.; Courivaud, C. Prevention of Post-Transplant Diabetes Mellitus: Towards a Personalized Approach. J. Pers. Med. 2022, 12, 116. https://doi.org/10.3390/jpm12010116
Ducloux D, Courivaud C. Prevention of Post-Transplant Diabetes Mellitus: Towards a Personalized Approach. Journal of Personalized Medicine. 2022; 12(1):116. https://doi.org/10.3390/jpm12010116
Chicago/Turabian StyleDucloux, Didier, and Cécile Courivaud. 2022. "Prevention of Post-Transplant Diabetes Mellitus: Towards a Personalized Approach" Journal of Personalized Medicine 12, no. 1: 116. https://doi.org/10.3390/jpm12010116