Pulmonary Manifestations of Plasma Cell Type Idiopathic Multicentric Castleman Disease: A Clinicopathological Study in Comparison with IgG4-Related Disease

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Case Selection
2.2. Diagnostic Criteria
2.3. Analysis of Clinical Features and CT Findings
2.4. Histological Evaluation
2.5. Immunohistochemistry
2.6. Statistical Analysis
2.7. Ethical Approval
3. Results
3.1. Clinical and Laboratory Findings
3.2. Evaluation of Chest CT Images
3.3. Analysis of Histological Features
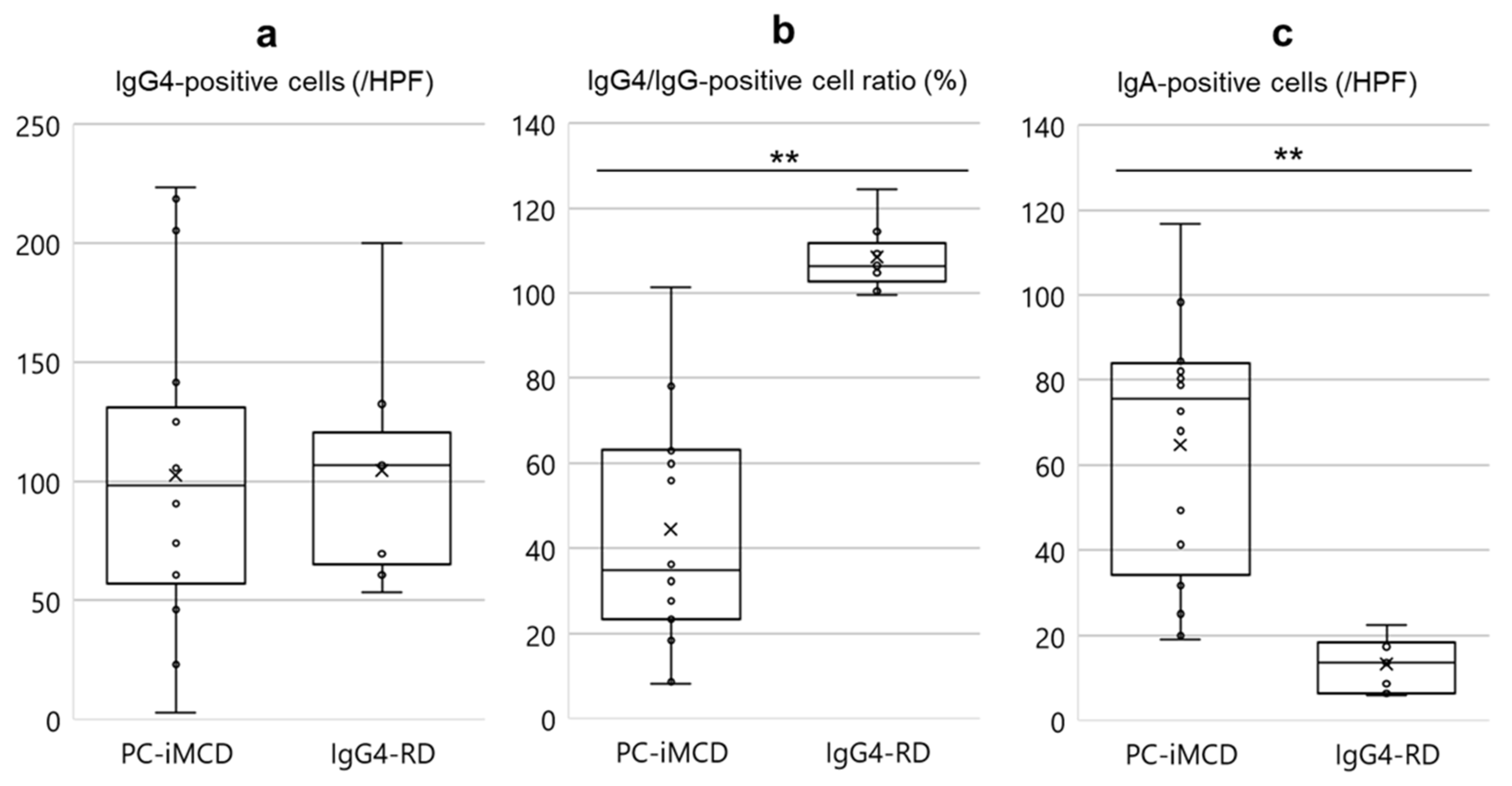
3.4. Analysis of Immunohistochemical Features
3.5. Analysis of Treatment and Clinical Course
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Castleman, B.; Iverson, L.; Menendez, V.P. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer 1956, 9, 822–830. [Google Scholar] [CrossRef]
- Yu, L.; Tu, M.; Cortes, J.; Xu-Monette, Z.Y.; Miranda, R.N.; Zhang, J.; Orlowski, R.Z.; Neelapu, S.; Boddu, P.C.; Akosile, M.A.; et al. Clinical and pathological characteristics of HIV- and HHV-8-negative Castleman disease. Blood 2017, 129, 1658–1668. [Google Scholar] [CrossRef] [Green Version]
- Frizzera, G.; Peterson, B.A.; Bayrd, E.D.; Goldman, A. A systemic lymphoproliferative disorder with morphologic features of Castleman’s disease: Clinical findings and clinicopathologic correlations in 15 patients. J. Clin. Oncol. 1985, 3, 1202–1216. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; Uldrick, T.S.; Bagg, A.; Frank, D.; Wu, D.; Srkalovic, G.; Simpson, D.; Liu, A.Y.; Menke, D.; Chandrakasan, S.; et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood 2017, 129, 1646–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fajgenbaum, D.C.; van Rhee, F.; Nabel, C.S. HHV-8-negative, idiopathic multicentric Castleman disease: Novel insights into biology, pathogenesis, and therapy. Blood 2014, 123, 2924–2933. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Pittaluga, S.; Jaffe, E.S. Multicentric Castleman disease: Where are we now? Semin. Diagn. Pathol. 2016, 33, 294–306. [Google Scholar] [CrossRef] [Green Version]
- Waterston, A.; Bower, M. Fifty years of multicentric Castleman’s disease. Acta Oncol. 2004, 43, 698–704. [Google Scholar] [CrossRef]
- Iyonaga, K.; Ichikado, K.; Muranaka, H.; Fujii, K.; Yamaguchi, T.; Suga, M. Multicentric Castleman’s disease manifesting in the lung: Clinical, radiographic, and pathologic findings and successful treatment with corticosteroid and cyclophosphamide. Intern. Med. 2003, 42, 182–186. [Google Scholar] [CrossRef] [Green Version]
- Bayes, H.K.; McInnes, I.B.; Chalmers, G.W. Castleman disease and lymphocytic interstitial pneumonia: A complex diagnostic and management challenge. Rheumatology 2017, 56, 665–668. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, V.; Zen, Y.; Chan, J.K.; Yi, E.E.; Sato, Y.; Yoshino, T.; Kloppel, G.; Heathcote, J.G.; Khosroshahi, A.; Ferry, J.A.; et al. Consensus statement on the pathology of IgG4-related disease. Mod. Pathol. 2012, 25, 1181–1192. [Google Scholar] [CrossRef] [Green Version]
- Kamisawa, T.; Zen, Y.; Pillai, S.; Stone, J.H. IgG4-related disease. Lancet 2015, 385, 1460–1471. [Google Scholar] [CrossRef]
- Stone, J.H.; Zen, Y.; Deshpande, V. IgG4-related disease. N. Engl. J. Med. 2012, 366, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Hamano, H.; Kawa, S.; Horiuchi, A.; Unno, H.; Furuya, N.; Akamatsu, T.; Fukushima, M.; Nikaido, T.; Nakayama, K.; Usuda, N.; et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N. Engl. J. Med. 2001, 344, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, T.; Kido, T.; Yatera, K.; Oda, K.; Kawanami, T.; Ishimoto, H.; Sakamoto, N.; Sano, A.; Yoshii, C.; Shimajiri, S.; et al. Assessment of pathologically diagnosed patients with Castleman’s disease associated with diffuse parenchymal lung involvement using the diagnostic criteria for IgG4-related disease. Lung 2013, 191, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kojima, M.; Takata, K.; Morito, T.; Asaoku, H.; Takeuchi, T.; Mizobuchi, K.; Fujihara, M.; Kuraoka, K.; Nakai, T.; et al. Systemic IgG4-related lymphadenopathy: A clinical and pathologic comparison to multicentric Castleman’s disease. Mod. Pathol. 2009, 22, 589–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rhee, F.; Voorhees, P.; Dispenzieri, A.; Fossa, A.; Srkalovic, G.; Ide, M.; Munshi, N.; Schey, S.; Streetly, M.; Pierson, S.K.; et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 2018, 132, 2115–2124. [Google Scholar] [CrossRef] [Green Version]
- Nishimoto, N.; Sasai, M.; Shima, Y.; Nakagawa, M.; Matsumoto, T.; Shirai, T.; Kishimoto, T.; Yoshizaki, K. Improvement in Castleman’s disease by humanized anti-interleukin-6 receptor antibody therapy. Blood 2000, 95, 56–61. [Google Scholar] [CrossRef]
- Nishimoto, N.; Kanakura, Y.; Aozasa, K.; Johkoh, T.; Nakamura, M.; Nakano, S.; Nakano, N.; Ikeda, Y.; Sasaki, T.; Nishioka, K.; et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood 2005, 106, 2627–2632. [Google Scholar] [CrossRef]
- Terasaki, Y.; Ikushima, S.; Matsui, S.; Hebisawa, A.; Ichimura, Y.; Izumi, S.; Ujita, M.; Arita, M.; Tomii, K.; Komase, Y.; et al. Comparison of clinical and pathological features of lung lesions of systemic IgG4-related disease and idiopathic multicentric Castleman’s disease. Histopathology 2017, 70, 1114–1124. [Google Scholar] [CrossRef]
- Otani, K.; Inoue, D.; Fujikura, K.; Komori, T.; Abe-Suzuki, S.; Tajiri, T.; Itoh, T.; Zen, Y. Idiopathic multicentric Castleman’s disease: A clinicopathologic study in comparison with IgG4-related disease. Oncotarget 2018, 9, 6691–6706. [Google Scholar] [CrossRef] [Green Version]
- Wallace, Z.S.; Naden, R.P.; Chari, S.; Choi, H.; Della-Torre, E.; Dicaire, J.F.; Hart, P.A.; Inoue, D.; Kawano, M.; Khosroshahi, A.; et al. The 2019 American College of Rheumatology/European League Against Rheumatism Classification Criteria for IgG4-Related Disease. Arthritis Rheumatol. 2020, 72, 7–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraya, T.; Ohkuma, K.; Fujiwara, M.; Miyaoka, C.; Wada, S.; Watanabe, T.; Mikura, S.; Inoue, M.; Oda, M.; Sada, M.; et al. Clinical characterization of 52 patients with immunoglobulin G4-related disease in a single tertiary center in Japan: Special reference to lung disease in thoracic high-resolution computed tomography. Respir. Med. 2017, 132, 62–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, D.; Zen, Y.; Abo, H.; Gabata, T.; Demachi, H.; Kobayashi, T.; Yoshikawa, J.; Miyayama, S.; Yasui, M.; Nakanuma, Y.; et al. Immunoglobulin G4-related lung disease: CT findings with pathologic correlations. Radiology 2009, 251, 260–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.T.; Lee, S.C.; Lu, C.C.; Tsai, C.L. Unusual presentation of Castleman’s disease mimicking lung cancer. Respirol. Case Rep. 2019, 7, e00416. [Google Scholar] [CrossRef]
- Haager, B.; Kayser, G.; Schmid, S.; Passlick, B.; Wiesemann, S. Intrapulmonary Castleman’s Disease Pretending to Be a Lung Cancer-Work Up of an Intrapulmonary Tumour. Ann. Thorac. Cardiovasc. Surg. 2016, 22, 258–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Kojima, M.; Takata, K.; Morito, T.; Mizobuchi, K.; Tanaka, T.; Inoue, D.; Shiomi, H.; Iwao, H.; Yoshino, T. Multicentric Castleman’s disease with abundant IgG4-positive cells: A clinical and pathological analysis of six cases. J. Clin. Pathol. 2010, 63, 1084–1089. [Google Scholar] [CrossRef]
- Han, Y.; Igawa, T.; Ogino, K.; Nishikori, A.; Gion, Y.; Yoshino, T.; Sato, Y. Hemosiderin deposition in lymph nodes of patients with plasma cell-type Castleman disease. J. Clin. Exp. Hematop. 2020, 60, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Manabe, A.; Igawa, T.; Takeuchi, M.; Gion, Y.; Yoshino, T.; Sato, Y. Immunohistochemical analysis of IgA expression differentiates IgG4-related disease from plasma cell-type Castleman disease. Med. Mol. Morphol. 2017, 50, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Post, G.R.; Bell, R.C.; Rjoop, A.; Lobo, R.H.; Yuan, Y.; Post, S.R. Diagnostic Utility of Interleukin-6 Expression by Immunohistochemistry in Differentiating Castleman Disease Subtypes and Reactive Lymphadenopathies. Ann. Clin. Lab. Sci. 2016, 46, 474–479. [Google Scholar]
- Satou, A.; Notohara, K.; Zen, Y.; Nakamura, S.; Yoshino, T.; Okazaki, K.; Sato, Y. Clinicopathological differential diagnosis of IgG4-related disease: A historical overview and a proposal of the criteria for excluding mimickers of IgG4-related disease. Pathol. Int. 2020, 70, 391–402. [Google Scholar] [CrossRef]
- Asano, N.; Sato, Y. Rheumatoid lymphadenopathy with abundant IgG4(+) plasma cells : A case mimicking IgG4-related disease. J. Clin. Exp. Hematop. 2012, 52, 57–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PC-iMCD (n = 16) | IgG4-RD (n = 7) | p-Value | ||
|---|---|---|---|---|
| Age (median ± SD) | 49.5 ± 12.0 | 70.0 ± 12.5 | 0.018 * | |
| Sex (M/F) | 7/9 | 7/0 | ― | |
| Symptom, n (%) | ||||
| Cough | 6 (37.5) | 1 (14.3) | 0.366 | |
| General fatigue | 5 (31.3) | 0 | 0.272 | |
| Fever | 4 (25.0) | 0 | 0.273 | |
| Shortness of breath | 3 (18.8) | 2 (28.6) | 0.621 | |
| Skin rash | 3 (18.8) | 0 | 0.526 | |
| Hemoptysis | 1 (6.3) | 0 | 1.000 | |
| Affected organs, n (%) | ||||
| Lung | 16 (100) | 7 (100) | ― | |
| Lymph nodes | 16 (100) | 5 (71.4) | 0.083 | |
| Skin | 3 (18.8) | 0 | 0.526 | |
| Spleen | 2 (12.5) | 0 | 1.000 | |
| Liver | 2 (12.5) | 0 | 1.000 | |
| Ureter | 1 (6.3) | 1 (14.3) | 0.526 | |
| Retroperitoneum | 0 | 1 (14.3) | 0.304 | |
| Kidney | 0 | 2 (28.6) | 0.083 | |
| Aorta(periaortitis) | 0 | 2 (28.6) | 0.083 | |
| Bile duct | 0 | 2 (28.6) | 0.083 | |
| Lacriminal gland | 0 | 2 (28.6) | 0.083 | |
| Pancreas | 0 | 3 (42.9) | 0.020 * | |
| Salivary gland | 1 (6.3) | 5 (71.4) | 0.003 * | |
| Laboratory findings (median ± SD) | ||||
| WBC (/μL) | 7800 ± 2059 | 7600 ± 1591 | 0.671 | |
| Hb (g/dL) | ||||
| Male | 13.0 ± 2.1 | 14.1 ± 1.7 | 0.275 | |
| Female | 11.1 ± 1.2 | ― | ― | |
| Plt (×104/µL) | 39.2 ± 9.6 | 21.4 ± 4.5 | <0.001 ** | |
| Alb (g/dL) | 3.0 ± 0.5 | 3.8 ± 0.7 | 0.019 * | |
| CRP (mg/dL) | 3.8 ± 3.7 | 0.3 ± 0.6 | <0.001 ** | |
| IgG (mg/dL) | 3883 ± 1242 | 2810 ± 1192 | 0.033 * | |
| IgG4 (mg/dL) | 374 ± 479 † | 1050 ± 590 | 0.018 * | |
| IgG4/IgG (%) | 11.3 ± 9.4 | 37.4 ± 12.8 | <0.001 ** | |
| IgA (mg/dL) | 567 ± 331 | 170 ± 74.8 ‡ | <0.001 ** | |
| IgE (IU/mL) | 1082 ± 870 † | 771 ± 1800 ‡ | 1.000 | |
| IL-6 (pg/mL) | 10.1 ± 8.5 † | ND | ― |
| PC-iMCD (n = 16) | IgG4-RD (n = 7) | p-Value | |
|---|---|---|---|
| Distribution pattern, n (%) | |||
| BvBs | 16 (100) | 7 (100) | NS |
| Interlobular septal | 15 (93.8) | 5 (71.4) | 0.209 |
| Alveolar interstitial | 7 (43.8) | 3 (42.9) | 1.000 |
| Nodular | 2 (12.5) | 1 (14.3) | 1.000 |
| Lymphoid follicles(/cm2) (median ± SD) | 74.0 ± 75.2 | 33.3 ± 37.2 | 0.053 |
| Sheet-like plasmacytosis, n (%) | 11 (68.8) | 1 (14.3) | 0.027 * |
| Fibrosis | |||
| Active fibrosis, n (%) | 10 (62.5) | 7 (100) | 0.124 |
| Dense hyalinized fibrosis, n (%) | 11 (68.8) | 3 (42.9) | 0.363 |
| Hemosiderin laden macrophages, n (%) | 2 (12.5) | 0 (0) | 1.000 |
| Obliterative vasculitis (/cm2) (median ± SD) | 13.3 ±16.1 | 20.8 ± 17.7 | 0.892 |
| Eosinophils (/HPF) (median ± SD) † | 10.0 ± 19.0 | 38.7 ± 24.2 | 0.004 * |
| Case Number | Age/Sex | Initial Therapy | Response to PSL | Additional Treatment | Outcome † | Observation Period (mo) |
|---|---|---|---|---|---|---|
| PC-iMCD | ||||||
| 1 | 46/M | PSL 10 mg | No response | Tocilizumab | remission | 83 |
| 2 | 40/F | PSL 40 mg | No response | Tocilizumab | remission | 93 |
| 3 | 53/M | PSL 30 mg | No response | Tocilizumab | remission | 90 |
| 4 | 44/M | PSL 30 mg | Repeatedly worsened during tapering | Tocilizumab | remission | 90 |
| 5 | 48/M | PSL 30 mg | No response | Tocilizumab | remission | 16 |
| 6 | 64/F | PSL 30 mg | No response | Tacrolimus | progression | 18 |
| 7 | 37/F | PSL 60 mg | No response | No additional treatment | progression | 46 |
| 8 | 41/F | PSL 20 mg | No response | No additional treatment | no change | 12 |
| 9 | 43/F | PSL 25 mg | Repeatedly worsened during tapering | No additional treatment | no change | 38 |
| 10 | 54/F | PSL 40 mg | Partial improvement | No additional treatment | partial remission | 180 |
| 11 | 60/M | PSL 40 mg | Partial improvement | No additional treatment | partial remission | 13 |
| 12 | 52/M | No treatment | ― | ― | progression | 88 |
| 13 | 51/M | No treatment | ― | ― | progression | 48 |
| 14 | 73/F | No treatment | ― | ― | remission | 36 |
| IgG4-RD | ||||||
| 1 | 39/M | PSL 40 mg | Improvement | No additional treatment | remission | 61 |
| 2 | 73/M | PSL 30 mg | Improvement | No additional treatment | remission | 88 |
| 3 | 70/M | PSL 40 mg | Improvement | No additional treatment | remission | 132 |
| 4 | 64/M | PSL 20 mg | Improvement | No additional treatment | remission | 132 |
| 5 | 71/M | PSL 20 mg | Improvement | No additional treatment | remission | 52 |
| 6 | 67/M | PSL 30 mg | Improvement | No additional treatment | remission | 31 |
| 7 | 77/M | No treatment | ― | No additional treatment | Dead | 2 |
| Suggestive of PC-iMCD | Suggestive of IgG4-RD | |
|---|---|---|
| Clinical Findings | Presence of fever or general fatigue | Presence of lesions in salivary gland or pancreas |
| CRP elevation of uncertain cause | ||
| Serum IgA elevation (above normal range) | ||
| Histological Findings | Sheet-like proliferation pattern of mature plasma cells | Eosinophils more than 20/HPF † |
| Immunohistochemical Findings | Strong expression of IL-6 immunostaining in interfollicular plasma cells and cells in germinal centers IgA-positive cells of more than 24/HPF † | Extremely high IgG4/IgG-positive cell ratio (>90%) † |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishimura, M.F.; Igawa, T.; Gion, Y.; Tomita, S.; Inoue, D.; Izumozaki, A.; Ubara, Y.; Nishimura, Y.; Yoshino, T.; Sato, Y. Pulmonary Manifestations of Plasma Cell Type Idiopathic Multicentric Castleman Disease: A Clinicopathological Study in Comparison with IgG4-Related Disease. J. Pers. Med. 2020, 10, 269. https://doi.org/10.3390/jpm10040269
Nishimura MF, Igawa T, Gion Y, Tomita S, Inoue D, Izumozaki A, Ubara Y, Nishimura Y, Yoshino T, Sato Y. Pulmonary Manifestations of Plasma Cell Type Idiopathic Multicentric Castleman Disease: A Clinicopathological Study in Comparison with IgG4-Related Disease. Journal of Personalized Medicine. 2020; 10(4):269. https://doi.org/10.3390/jpm10040269
Chicago/Turabian StyleNishimura, Midori Filiz, Takuro Igawa, Yuka Gion, Sakura Tomita, Dai Inoue, Akira Izumozaki, Yoshifumi Ubara, Yoshito Nishimura, Tadashi Yoshino, and Yasuharu Sato. 2020. "Pulmonary Manifestations of Plasma Cell Type Idiopathic Multicentric Castleman Disease: A Clinicopathological Study in Comparison with IgG4-Related Disease" Journal of Personalized Medicine 10, no. 4: 269. https://doi.org/10.3390/jpm10040269