
A Novel Two-Gene Expression-Based Prognostic Score in Malignant Pleural Mesothelioma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Model Building
2.3. Gene Set Enrichment Analysis
2.4. Cibersort
2.5. Drug Sensitivity Analysis
2.6. Common Statistical Procedures
3. Results
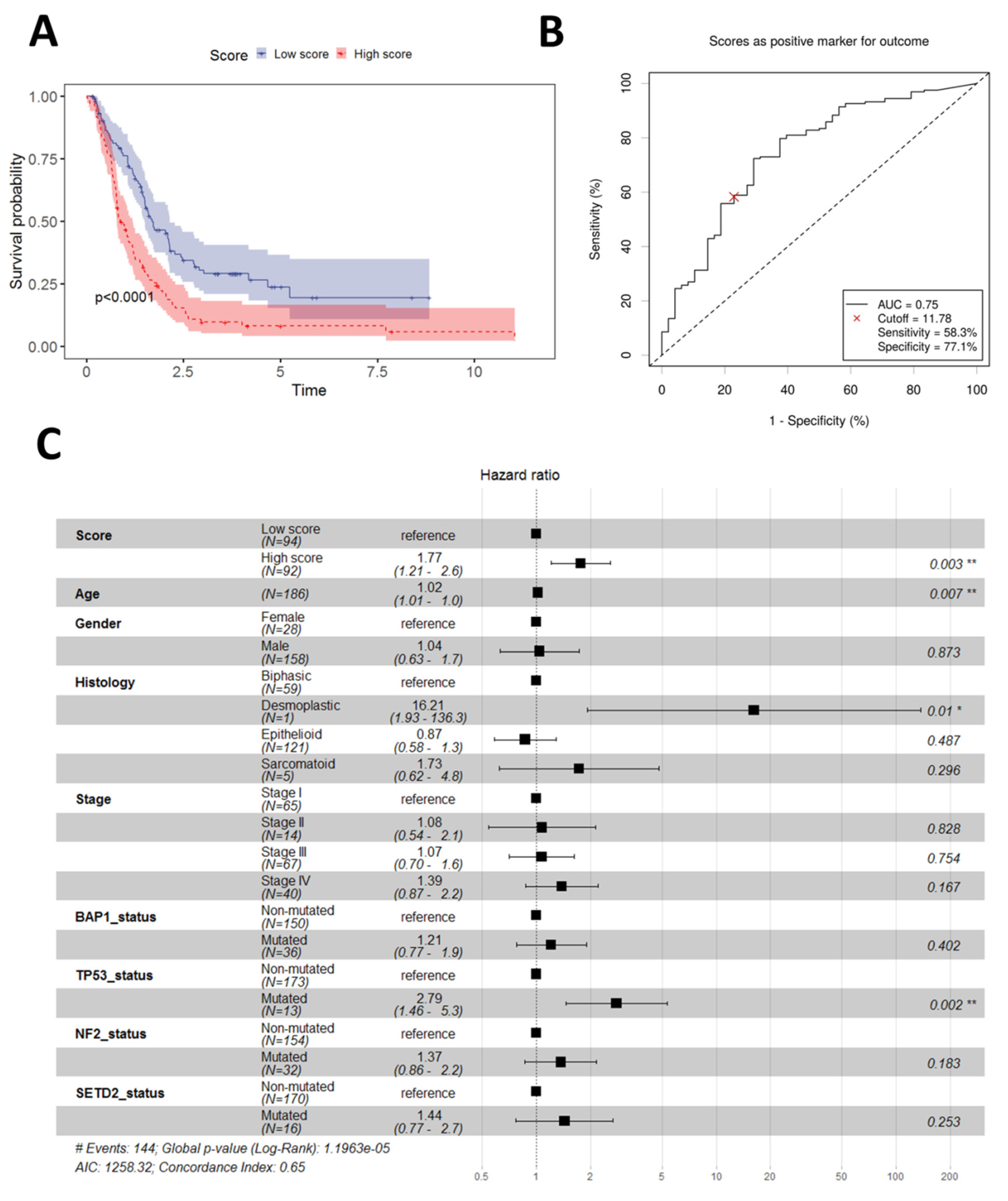
3.1. Building and Initial Performance of a Two-Gene Prognostic Score (2-PS)
3.2. Validation of the 2-PS
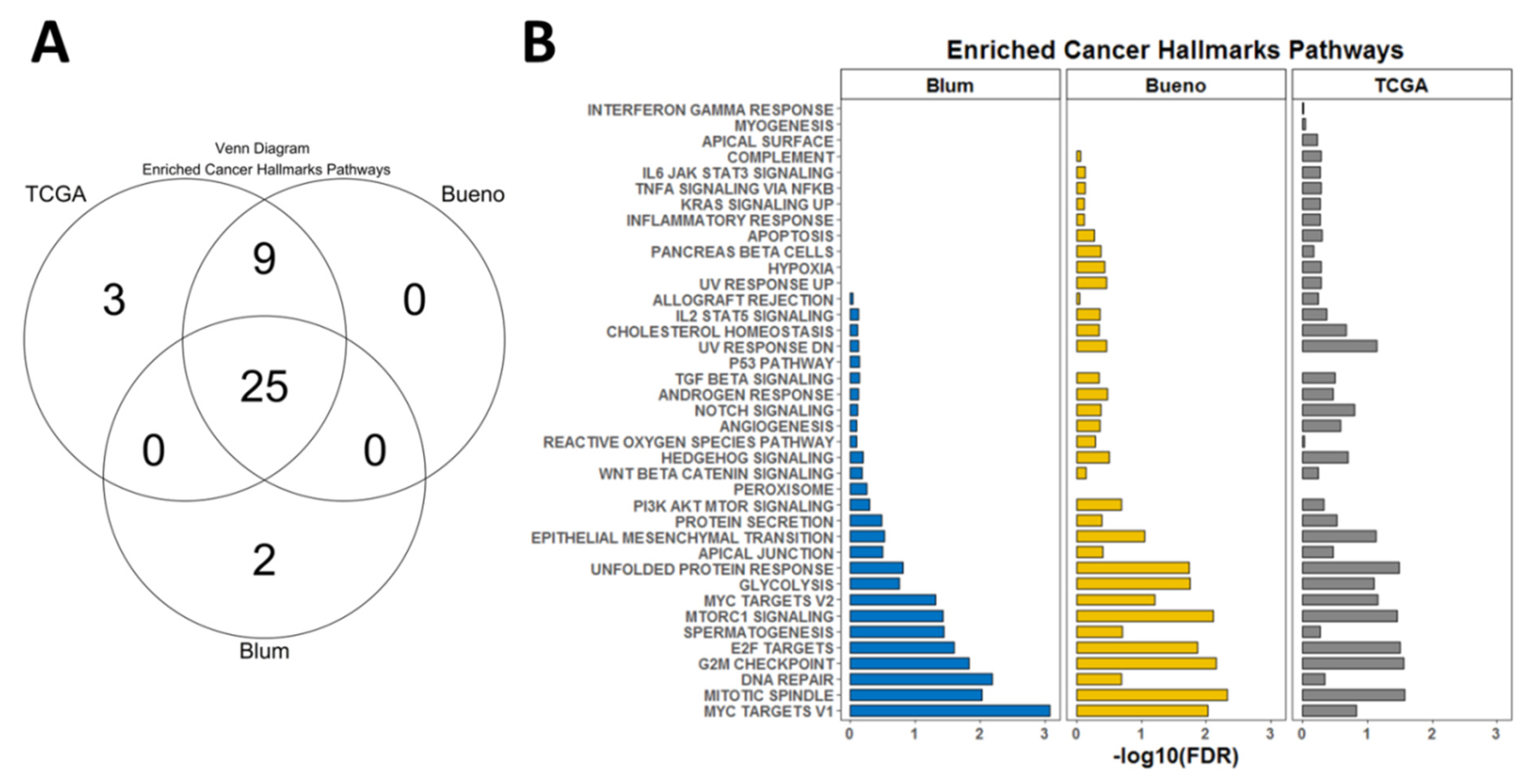
3.3. Gene Set Enrichment Analysis
3.4. Correlation with Immune Signatures
3.5. Potential Predictive Power of the 2-PS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alpert, N.; van Gerwen, M.; Taioli, E. Epidemiology of mesothelioma in the 21st century in Europe and the United States, 40 years after restricted/banned asbestos use. Transl. Lung Cancer Res. 2020, 9 (Suppl. 1), S28. [Google Scholar] [CrossRef]
- Shao, C.; Li, G.; Huang, L.; Pruitt, S.; Castellanos, E.; Frampton, G.; Carson, K.R.; Snow, T.; Singal, G.; Fabrizio, D. Prevalence of high tumor mutational burden and association with survival in patients with less common solid tumors. JAMA Netw. Open 2020, 3, e2025109. [Google Scholar] [CrossRef]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A. Integrative molecular characterization of malignant pleural mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef]
- Fennell, D.A.; Ewings, S.; Ottensmeier, C.; Califano, R.; Hanna, G.G.; Hill, K.; Danson, S.; Steele, N.; Nye, M.; Johnson, L. Nivolumab versus placebo in patients with relapsed malignant mesothelioma (CONFIRM): A multicentre, double-blind, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 1530–1540. [Google Scholar] [CrossRef]
- Peters, S.; Scherpereel, A.; Cornelissen, R.; Oulkhouir, Y.; Greillier, L.; Kaplan, M.; Talbot, T.; Monnet, I.; Hiret, S.; Baas, P. First-line nivolumab plus ipilimumab versus chemotherapy in patients with unresectable malignant pleural mesothelioma: 3-year outcomes from CheckMate 743. Ann. Oncol. 2022, 33, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Nakagawa, K.; Fujimoto, N.; Kuribayashi, K.; Guren, T.K.; Calabrò, L.; Shapira-Frommer, R.; Gao, B.; Kao, S.; Matos, I. Efficacy and safety of pembrolizumab in patients with advanced mesothelioma in the open-label, single-arm, phase 2 KEYNOTE-158 study. Lancet Respir. Med. 2021, 9, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Wadowski, B.; De Rienzo, A.; Bueno, R. The molecular basis of malignant pleural mesothelioma. Thorac. Surg. Clin. 2020, 30, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef]
- Blum, Y.; Meiller, C.; Quetel, L.; Elarouci, N.; Ayadi, M.; Tashtanbaeva, D.; Armenoult, L.; Montagne, F.; Tranchant, R.; Renier, A. Dissecting heterogeneity in malignant pleural mesothelioma through histo-molecular gradients for clinical applications. Nat. Commun. 2019, 10, 1333. [Google Scholar] [CrossRef]
- Yu, C.; Mannan, A.M.; Yvone, G.M.; Ross, K.N.; Zhang, Y.-L.; Marton, M.A.; Taylor, B.R.; Crenshaw, A.; Gould, J.Z.; Tamayo, P. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat. Biotechnol. 2016, 34, 419–423. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M. Defining a cancer dependency map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [PubMed]
- Boehm, J.S.; Golub, T.R. An ecosystem of cancer cell line factories to support a cancer dependency map. Nat. Rev. Genet. 2015, 16, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Yu, A.; Kim, S.; Kang, J.; Hong, S.M. Robust likelihood-based survival modeling with microarray data. J. Stat. Softw. 2009, 29, 1–16. [Google Scholar] [CrossRef]
- Budczies, J.; Klauschen, F.; Sinn, B.V.; Gyorffy, B.; Schmitt, W.D.; Darb-Esfahani, S.; Denkert, C. Cutoff Finder: A comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS ONE 2012, 7, e51862. [Google Scholar] [CrossRef]
- Shivarov, V.; Dolnik, A.; Lang, K.M.; Krönke, J.; Kuchenbauer, F.; Paschka, P.; Gaidzik, V.I.; Döhner, H.; Schlenk, R.F.; Döhner, K. MicroRNA expression-based outcome prediction in acute myeloid leukemia: Novel insights through cross-platform integrative analyses. Haematologica 2016, 101, e454. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Yang, T.-H.O.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A. The immune landscape of cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H. A landscape of pharmacogenomic interactions in cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef]
- Mandrekar, J.N. Receiver operating characteristic curve in diagnostic test assessment. J. Thorac. Oncol. 2010, 5, 1315–1316. [Google Scholar] [CrossRef]
- Pass, H.I.; Liu, Z.; Wali, A.; Bueno, R.; Land, S.; Lott, D.; Siddiq, F.; Lonardo, F.; Carbone, M.; Draghici, S. Gene expression profiles predict survival and progression of pleural mesothelioma. Clin. Cancer Res. 2004, 10, 849–859. [Google Scholar] [CrossRef]
- De Reynies, A.; Jaurand, M.-C.; Renier, A.; Couchy, G.; Hysi, I.; Elarouci, N.; Galateau-Sallé, F.; Copin, M.-C.; Hofman, P.; Cazes, A. Molecular classification of malignant pleural mesothelioma: Identification of a poor prognosis subgroup linked to the epithelial-to-mesenchymal transition. Clin. Cancer Res. 2014, 20, 1323–1334. [Google Scholar] [CrossRef]
- Gordon, G.J.; Jensen, R.V.; Hsiao, L.-L.; Gullans, S.R.; Blumenstock, J.E.; Richards, W.G.; Jaklitsch, M.T.; Sugarbaker, D.J.; Bueno, R. Using gene expression ratios to predict outcome among patients with mesothelioma. J. Natl. Cancer Inst. 2003, 95, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.J.; Rockwell, G.N.; Jensen, R.V.; Rheinwald, J.G.; Glickman, J.N.; Aronson, J.P.; Pottorf, B.J.; Nitz, M.D.; Richards, W.G.; Sugarbaker, D.J. Identification of novel candidate oncogenes and tumor suppressors in malignant pleural mesothelioma using large-scale transcriptional profiling. Am. J. Pathol. 2005, 166, 1827–1840. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.J.; Dong, L.; Yeap, B.Y.; Richards, W.G.; Glickman, J.N.; Edenfield, H.; Mani, M.; Colquitt, R.; Maulik, G.; Van Oss, B. Four-gene expression ratio test for survival in patients undergoing surgery for mesothelioma. J. Natl. Cancer Inst. 2009, 101, 678–686. [Google Scholar] [CrossRef]
- Zhou, J.-G.; Zhong, H.; Zhang, J.; Jin, S.-H.; Roudi, R.; Ma, H. Development and validation of a prognostic signature for malignant pleural mesothelioma. Front. Oncol. 2019, 9, 78. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, X.; Hou, J.; Geng, L.; Liang, X.; Ruan, Z.; Guo, H.; Nan, K.; Jiang, L. Identification of a five-gene signature for predicting survival in malignant pleural mesothelioma patients. Front. Genet. 2020, 11, 899. [Google Scholar] [CrossRef]
- López-Ríos, F.; Chuai, S.; Flores, R.; Shimizu, S.; Ohno, T.; Wakahara, K.; Illei, P.B.; Hussain, S.; Krug, L.; Zakowski, M.F. Global gene expression profiling of pleural mesotheliomas: Overexpression of aurora kinases and P16/CDKN2A deletion as prognostic factors and critical evaluation of microarray-based prognostic prediction. Cancer Res. 2006, 66, 2970–2979. [Google Scholar] [CrossRef]
- Musacchio, A.; Hardwick, K.G. The spindle checkpoint: Structural insights into dynamic signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 731–741. [Google Scholar] [CrossRef]
- Morani, F.; Bisceglia, L.; Rosini, G.; Mutti, L.; Melaiu, O.; Landi, S.; Gemignani, F. Identification of overexpressed genes in malignant pleural mesothelioma. Int. J. Mol. Sci. 2021, 22, 2738. [Google Scholar] [CrossRef] [PubMed]
- Suraokar, M.; Nunez, M.; Diao, L.; Chow, C.; Kim, D.; Behrens, C.; Lin, H.; Lee, S.; Raso, G.; Moran, C. Expression profiling stratifies mesothelioma tumors and signifies deregulation of spindle checkpoint pathway and microtubule network with therapeutic implications. Ann. Oncol. 2014, 25, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Busacca, S.; O’Regan, L.; Singh, A.; Sharkey, A.J.; Dawson, A.G.; Dzialo, J.; Parsons, A.; Kumar, N.; Schunselaar, L.M.; Guppy, N. BRCA1/MAD2L1 Deficiency Disrupts the Spindle Assembly Checkpoint to Confer Vinorelbine Resistance in MesotheliomaBRCA1/MAD2L1 Expression Predicts Response to Vinorelbine. Mol. Cancer Ther. 2021, 20, 379–388. [Google Scholar] [CrossRef]
- Conchon, S.; Cao, X.; Barlowe, C.; Pelham, H.R. Got1p and Sft2p: Membrane proteins involved in traffic to the Golgi complex. EMBO J. 1999, 18, 3934–3946. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, V.; Naef, F.; Rao, P.H.; Reuter, V.; Mok, S.C.; Bosl, G.J.; Koul, S.; Murty, V.V.; Kucherlapati, R.S.; Chaganti, R. Genomic and expression analysis of the 12p11-p12 amplicon using EST arrays identifies two novel amplified and overexpressed genes. Cancer Res. 2002, 62, 6218–6223. [Google Scholar]
- Liu, T.; Liu, B.; Liu, Y.; Feng, X.; Jiang, X.; Long, J.; Gao, Q.; Yang, Z. Vesicle transporter GOLT1B mediates the cell membrane localization of DVL2 and PD-L2 and promotes colorectal cancer metastasis. Cancer Cell Int. 2021, 21, 287. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, W.; Cai, W.; Chen, Y.; Cai, X.; Tang, D.; Tang, M.; Dai, Y. Multi-omics analyses revealed GOLT1B as a potential prognostic gene in breast cancer probably regulating the immune microenvironment. Front. Oncol. 2022, 11, 5871. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Lee, H.-S.; Burt, B.M.; Amos, C.I.; Cheng, C. A combination of intrinsic and extrinsic features improves prognostic prediction in malignant pleural mesothelioma. Br. J. Cancer 2022, 127, 1691–1700. [Google Scholar] [CrossRef]
- Fournel, L.; Charrier, T.; Huriet, M.; Iaffaldano, A.; Lupo, A.; Damotte, D.; Arrondeau, J.; Alifano, M. Prognostic impact of inflammation in malignant pleural mesothelioma: A large-scale analysis of consecutive patients. Lung Cancer 2022, 166, 221–227. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shivarov, V.; Blazhev, G.; Yordanov, A. A Novel Two-Gene Expression-Based Prognostic Score in Malignant Pleural Mesothelioma. Diagnostics 2023, 13, 1556. https://doi.org/10.3390/diagnostics13091556
Shivarov V, Blazhev G, Yordanov A. A Novel Two-Gene Expression-Based Prognostic Score in Malignant Pleural Mesothelioma. Diagnostics. 2023; 13(9):1556. https://doi.org/10.3390/diagnostics13091556
Chicago/Turabian StyleShivarov, Velizar, Georgi Blazhev, and Angel Yordanov. 2023. "A Novel Two-Gene Expression-Based Prognostic Score in Malignant Pleural Mesothelioma" Diagnostics 13, no. 9: 1556. https://doi.org/10.3390/diagnostics13091556