
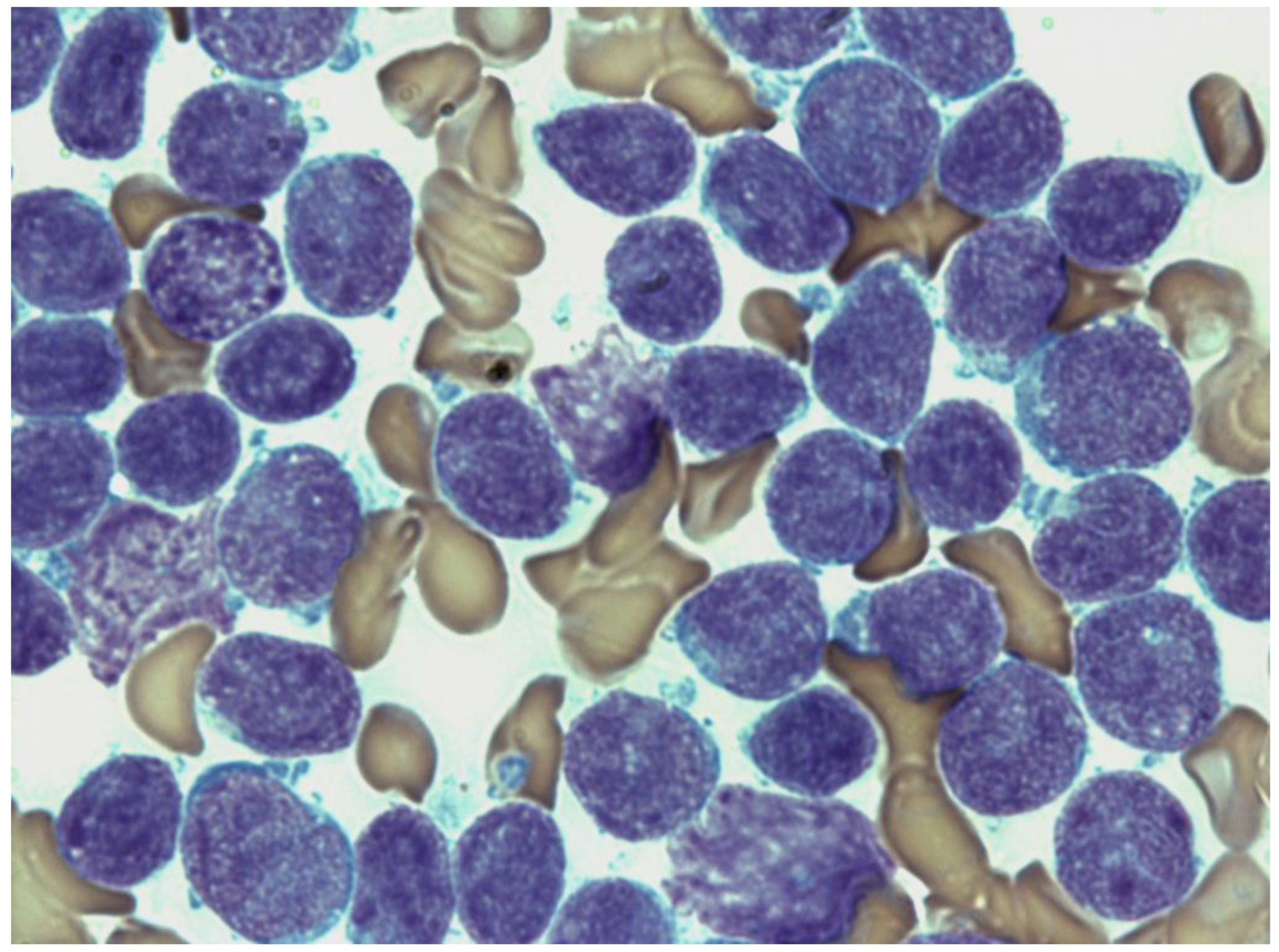
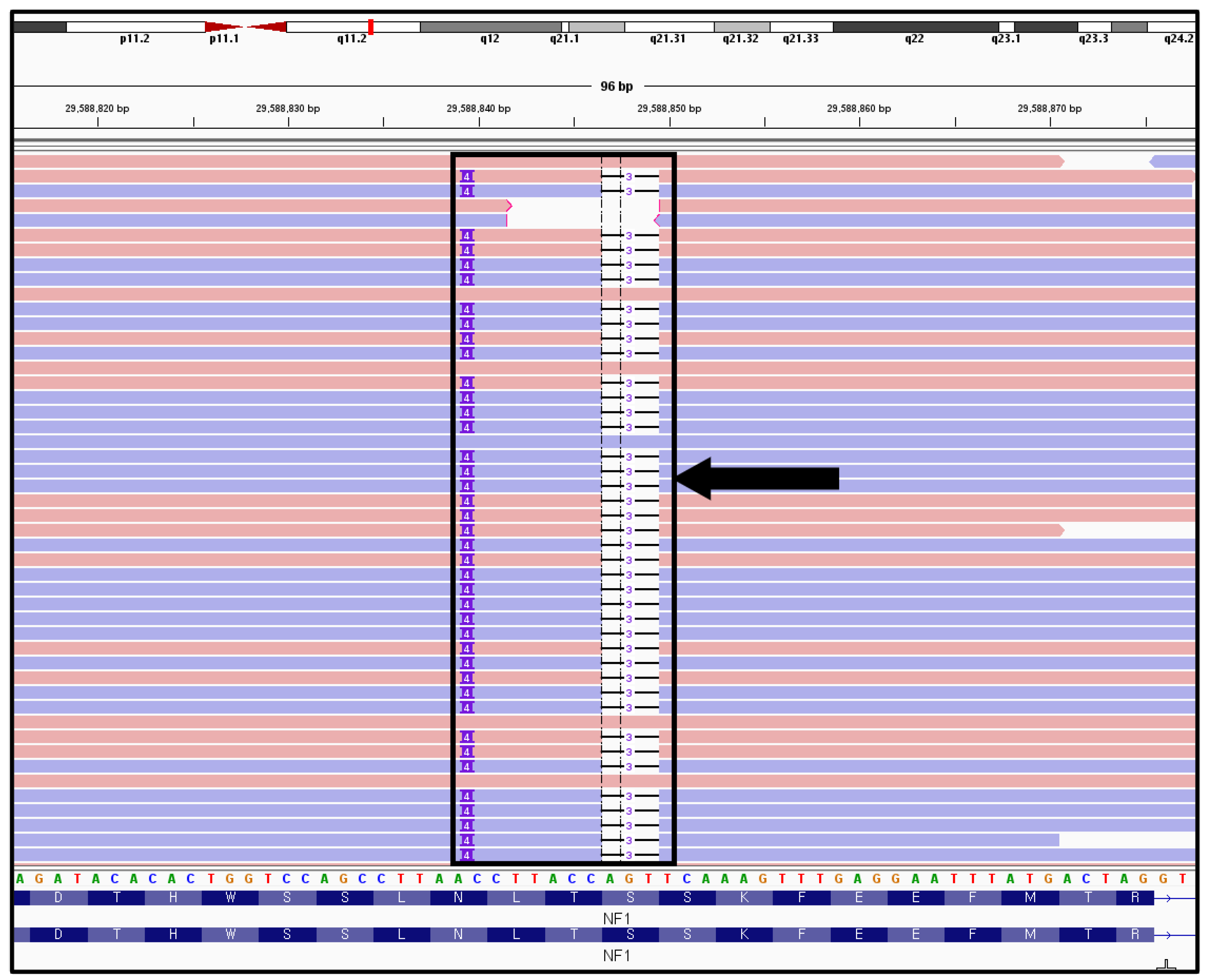
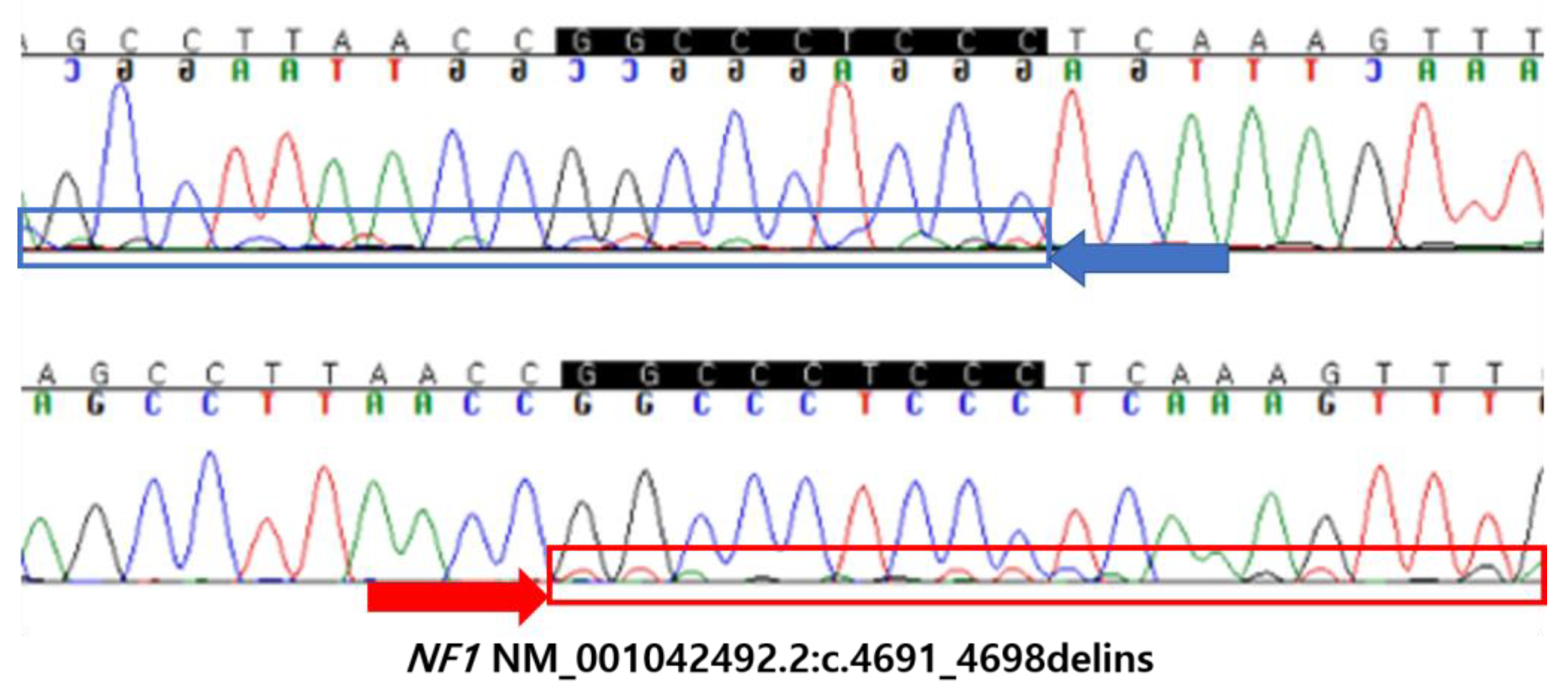
Neurofibromatosis Symptom-Lacking B-Cell Lineage Acute Lymphoblastic Leukemia with Only an NF1 Gene Pathogenic Variant

{kind=link}
{kind=link}
{kind=link}
Abstract
:Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pui, C.-H.; Schrappe, M.; Ribeiro, R.C.; Niemeyer, C.M. Childhood and Adolescent Lymphoid and Myeloid Leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2004, 2004, 118–145. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Iacobucci, I.; Mullighan, C.G. Genetic Basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Malard, F.; Mohty, M. Acute lymphoblastic leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef] [PubMed]
- Parkin, B.; Ouillette, P.; Wang, Y.; Liu, Y.; Wright, W.; Roulston, D.; Purkayastha, A.; Dressel, A.; Karp, J.; Bockenstedt, P.; et al. NF1 inactivation in adult acute myelogenous leukemia. Clin. Cancer Res. 2010, 16, 4135–4147. [Google Scholar] [CrossRef] [PubMed]
- Balgobind, B.V.; Van Vlierberghe, P.; van den Ouweland, A.M.; Beverloo, H.B.; Terlouw-Kromosoeto, J.N.; van Wering, E.R.; Meijerink, J.P. Leukemia-associated NF1 inactivation in patients with pediatric T-ALL and AML lacking evidence for neurofibromatosis. Blood 2008, 111, 4322–4328. [Google Scholar] [CrossRef]
- Eisfeld, A.K.; Kohlschmidt, J.; Mrózek, K.; Mims, A.; Walker, C.J.; Blachly, J.S.; Bloomfield, C.D. NF1 mutations are recurrent in adult acute myeloid leukemia and confer poor outcome. Leukemia 2018, 32, 2536–2545. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Nikiforova, M.N. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Prim. 2017, 3, 17004. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Forbes, S.A. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D977. [Google Scholar] [CrossRef]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Kattman, B.L. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Yeung, D.T.O.; Osborn, M.P.; White, D.L. B-cell acute lymphoblastic leukaemia: Recent discoveries in molecular pathology, their prognostic significance, and a review of the current classification. Br. J. Haematol. 2022, 197, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Holmfeldt, L.; Wei, L.; Diaz-Flores, E.; Walsh, M.; Zhang, J.; Ding, L.; Mullighan, C.G. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Mullally, A.; Ebert, B.L. NF1 inactivation revs up Ras in adult acute myelogenous leukemia. Clin. Cancer Res. 2010, 16, 4074–4076. [Google Scholar] [CrossRef] [PubMed]
- Oshrine, B.; Grana, N.; Moore, C.; Nguyen, J.; Crenshaw, M.; Edwards, M.; Tabori, U. B-cell acute lymphoblastic leukemia with high mutation burden presenting in a child with constitutional mismatch repair deficiency. Blood Adv. 2019, 3, 1795–1798. [Google Scholar] [CrossRef] [PubMed]
- Boissel, N.; Baruchel, A. Acute lymphoblastic leukemia in adolescent and young adults: Treat as adults or as children? Blood 2018, 132, 351–361. [Google Scholar] [CrossRef]
- Lafage-Pochitaloff, M.; Baranger, L.; Hunault, M.; Cuccuini, W.; Lefebvre, C.; Bidet, A.; Dombret, H. Impact of cytogenetic abnormalities in adults with Ph-negative B-cell precursor acute lymphoblastic leukemia. Blood 2017, 130, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Ofran, Y.; Izraeli, S. BCR-ABL (Ph)-like acute leukemia-Pathogenesis, diagnosis and therapeutic options. Blood Rev. 2017, 31, 11–16. [Google Scholar] [CrossRef]
- Tasian, S.K.; Loh, M.L.; Hunger, S.P. Philadelphia chromosome-like acute lymphoblastic leukemia. Blood 2017, 130, 2064–2072. [Google Scholar] [CrossRef]
- Kaburagi, T.; Yamato, G.; Shiba, N.; Yoshida, K.; Hara, Y.; Tabuchi, K.; Hayashi, Y. Clinical significance of RAS pathway alterations in pediatric acute myeloid leukemia. Haematologica 2021, 107, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.F.; Braun, B.S.; Shannon, K.M. Targeting oncogenic Ras signaling in hematologic malignancies. Blood 2012, 120, 3397–3406. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Grupp, S.A. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; Mullighan, C.G. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Ganesan, P.; Veeramani, R.; Kumar, V.D. Philadelphia-Like Acute Lymphoblastic Leukemia: A Systematic Review. Clin. Lymphoma Myeloma Leuk. 2021, 21, e57–e65. [Google Scholar] [CrossRef]
- De Vos, N.; Hofmans, M.; Lammens, T.; De Wilde, B.; Van Roy, N.; De Moerloose, B. Targeted therapy in juvenile myelomonocytic leukemia: Where are we now? Pediatr. Blood Cancer 2022, 69, e29930. [Google Scholar] [CrossRef]
- Ney, G.M.; McKay, L.; Koschmann, C.; Mody, R.; Li, Q. The Emerging Role of Ras Pathway Signaling in Pediatric Cancer. Cancer Res. 2020, 80, 5155–5163. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.-J.; Jang, H. Anticancer drug resistance: An update and perspective. Drug Resist. Updates 2021, 59, 100796. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Z.; Lee, J.H. Neurofibromatosis Symptom-Lacking B-Cell Lineage Acute Lymphoblastic Leukemia with Only an NF1 Gene Pathogenic Variant. Diagnostics 2023, 13, 1486. https://doi.org/10.3390/diagnostics13081486
Kim Z, Lee JH. Neurofibromatosis Symptom-Lacking B-Cell Lineage Acute Lymphoblastic Leukemia with Only an NF1 Gene Pathogenic Variant. Diagnostics. 2023; 13(8):1486. https://doi.org/10.3390/diagnostics13081486
Chicago/Turabian StyleKim, Zehwan, and Jong Ho Lee. 2023. "Neurofibromatosis Symptom-Lacking B-Cell Lineage Acute Lymphoblastic Leukemia with Only an NF1 Gene Pathogenic Variant" Diagnostics 13, no. 8: 1486. https://doi.org/10.3390/diagnostics13081486