

Omphalocele and Cardiac Abnormalities—The Importance of the Association

 , , , , , and
, , , , , and
Abstract
:1. Introduction
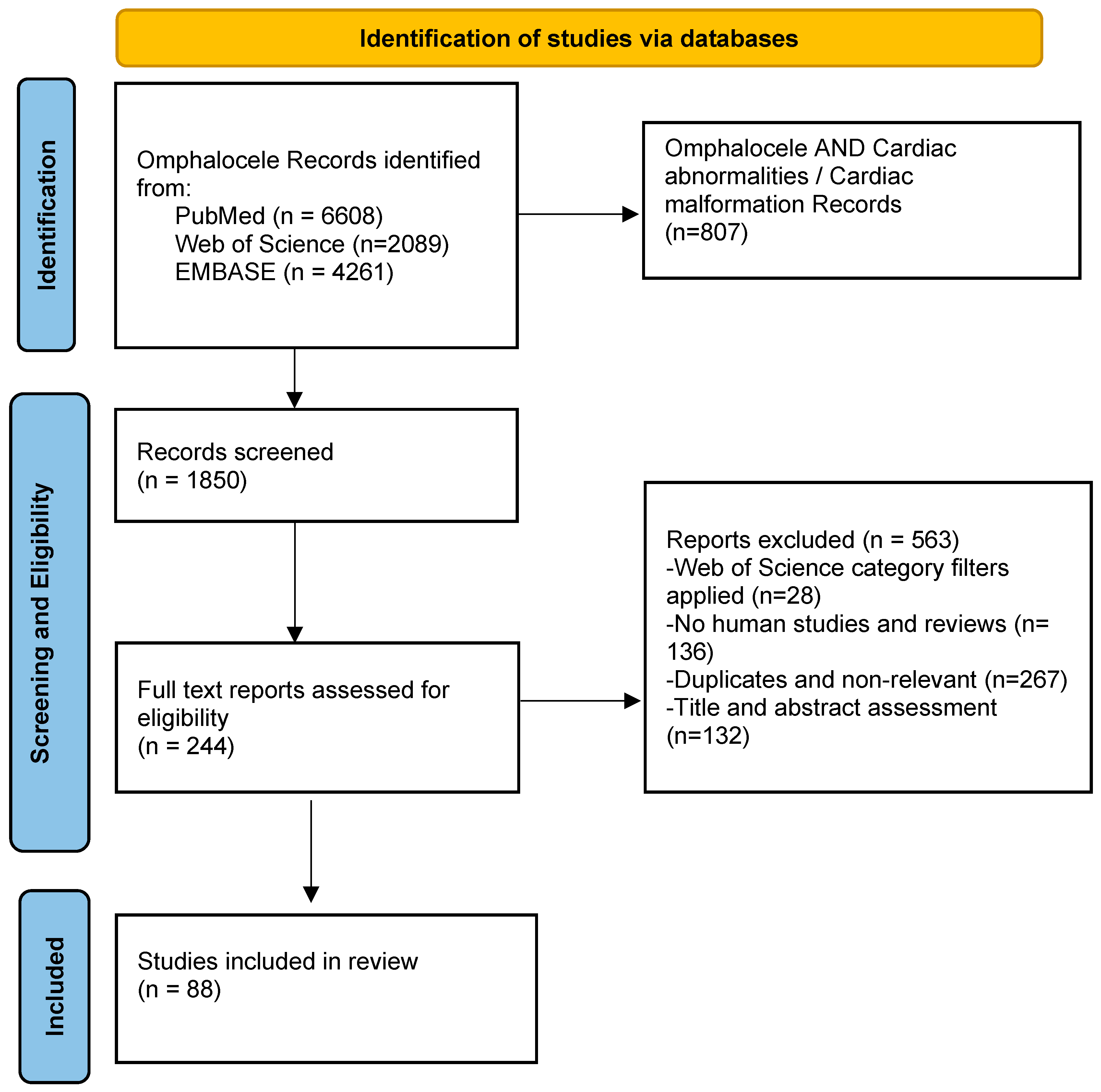
2. Materials and Methods
Electronic Databases and Search Strategy
3. Results
Literature Review
4. Epidemiology
5. Omphalocele Patients with Cardiac Abnormalities
5.1. Atrial Septal Defects (ASD)
5.2. Ventricular Septal Defect (VSD)
5.3. Hypoplastic Left Heart Syndrome
5.4. Tricuspid Atresia (TA)
5.5. Ectopia Cordis (EC)
5.6. Associated Abnormalities of Systemic Veins
5.7. Pulmonary Hypertension and Right Ventricular Dysfunction
6. Chromosomal Abnormalities and Syndromes
6.1. PAGOD Syndrome
6.2. Beckwith–Wiedemann Syndrome (BWS)
6.3. Pentalogy of Cantrell (PC)
6.4. Left Atrial Isomerism (LAI)
6.5. OEIS Syndrome
6.6. Limb Body Wall Complex (LBWC)
6.7. The VACTERL Malformation Association
6.8. The ADAM Sequence (Amniotic Deformitis, Adhesions, Mutilations)
6.9. Otopalatodigital Spectrum Disorders
7. Management
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mai, C.T.; Isenburg, J.L.; Canfield, M.A.; Meyer, R.E.; Correa, A.; Alverson, C.J.; Lupo, P.J.; Riehle-Colarusso, T.; Cho, S.J.; Aggarwal, D.; et al. National population-based estimates for major birth defects, 2010–2014. Birth Defects Res. 2019, 111, 1420–1435. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.; Salemi, J.L.; Tanner, J.P.M.; Ramakrishnan, R.M.; Feldkamp, M.L.P.; Marengo, L.K.M.; Meyer, R.E.; Druschel, C.M.M.; Rickard, R.M.; Kirby, R.S.P. Prevalence, Correlates, and Outcomes of Omphalocele in the United States, 1995–2005. Obstet. Gynecol. 2015, 126, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Fleurke-Rozema, H.; van de Kamp, K.; Bakker, M.; Pajkrt, E.; Bilardo, C.; Snijders, R. Prevalence, timing of diagnosis and pregnancy outcome of abdominal wall defects after the introduction of a national prenatal screening program. Prenat. Diagn. 2017, 37, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Țarcă, V.; Țarcă, E.; Luca, F.-A. The Impact of the Main Negative Socio-Economic Factors on Female Fertility. Healthcare 2022, 10, 734. [Google Scholar] [CrossRef] [PubMed]
- Snijders, R.J.; Sebire, N.J.; Souka, A.; Santiago, C.; Nicolaides, K.H. Fetal exomphalos and cromosomal defects: Relationship to maternal age and gestation. Ultrasound Obstet. Gynecol. 1995, 6, 250–255. [Google Scholar] [CrossRef]
- Ayub, S.S.; Taylor, J.A. Cardiac anomalies associated with omphalocele. Semin. Pediatr. Surg. 2019, 28, 111–114. [Google Scholar] [CrossRef]
- Oluwafemi, O.O.; Benjamin, R.H.; Sanchez, M.L.N.; Scheuerle, A.E.; Schaaf, C.P.; Mitchell, L.E.; Langlois, P.H.; Canfield, M.A.; Swartz, M.D.; Scott, D.A.; et al. Birth defects that co-occur with non-syndromic gastroschisis and omphalocele. Am. J. Med. Genet. A 2020, 182, 2581–2593. [Google Scholar] [CrossRef]
- Tarcă, E.; Aprodu, S. Past and present in omphalocele treatment in Romania. Chirurgia 2014, 109, 507–513. [Google Scholar]
- Kumar, H.R.; Jester, A.L.; Ladd, A.P. Impact of omphalocele size on associated conditions. J. Pediatr. Surg. 2008, 43, 2216–2219. [Google Scholar] [CrossRef]
- Duhamel, B. Embriology of exomphalos and allied malformations. Arch. Dis. Child. 1963, 38, 142–147. [Google Scholar] [CrossRef]
- Gross, R.E.; Blodgett, J.B. Omphalocele (umbilical eventration) in the newly born. Surg. Gynecol. Obst. 1940, 71, 520. [Google Scholar]
- Brewer, S.; Williams, T. Loss of AP-2α impacts multiple aspects of ventral body wall development and closure. Dev. Biol. 2004, 267, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.A.; Hashmi, A.; Islam, S. Insights into embryology and development of omphalocele. Semin. Pediatr. Surg. 2019, 28, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Brantberg, A.; Blaas, H.-G.K.; Haugen, S.E.; Eik-Nes, S.H. Characteristics and outcome of 90 cases of fetal omphalocele. Ultrasound Obstet. Gynecol. 2005, 26, 527–537. [Google Scholar] [CrossRef]
- Ma, X.; Adelstein, R.S. A Point Mutation in Myh10 Causes Major Defects in Heart Development and Body Wall Closure. Circ. Cardiovasc. Genet. 2014, 7, 257–265, Erratum in Circ. Cardiovasc. Genet. 2014, 7, 570. [Google Scholar] [CrossRef]
- Anyanwu, L.-J.C.; Ade-Ajayi, N.; Rolle, U. Major abdominal wall defects in the low- and middle-income setting: Current status and priorities. Pediatr. Surg. Int. 2020, 36, 579–590. [Google Scholar] [CrossRef]
- Bohîlțea, R.E.; Dima, V.; Ducu, I.; Iordache, A.M.; Mihai, B.M.; Munteanu, O.; Grigoriu, C.; Veduță, A.; Pelinescu-Onciul, D.; Vlădăreanu, R. Clinically Relevant Prenatal Ultrasound Diagnosis of Umbilical Cord Pathology. Diagnostics 2022, 12, 236. [Google Scholar] [CrossRef]
- Ţarcă, E.; Cojocaru, E.; Trandafir, L.M.; Luca, A.C.; Tiutiucă, R.C.; Butnariu, L.I.; Costea, C.F.; Radu, I.; Moscalu, M.; Ţarcă, V. Current Challenges in the Treatment of the Omphalocele—Experience of a Tertiary Center from Romania. J. Clin. Med. 2022, 11, 5711. [Google Scholar] [CrossRef]
- Verla, M.A.; Style, C.C.; Olutoye, O.O. Prenatal diagnosis and management of omphalocele. Semin. Pediatr. Surg. 2019, 28, 84–88. [Google Scholar] [CrossRef]
- Boulton, S.L.; McKenna, D.S.; Cly, G.C.; Webb, D.C.; Bantz, J.; Sonek, J. Cardiac axis in fetuses with abdominal wall defects. Ultrasound Obstet. Gynecol. 2006, 28, 785–788. [Google Scholar] [CrossRef]
- Heider, A.L.; Strauss, R.A.; Kuller, J.A. Omphalocele: Clinical outcomes in cases with normal karyotypes. Am. J. Obstet. Gynecol. 2004, 190, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.; Aldrink, J.H.; Richards, B.; Corbitt, R.; Stiver, C.; Cua, C.L. Usefulness of Postnatal Echocardiograms in Patients with Omphaloceles Who Previously Had a Normal Fetal Echocardiogram. Cardiol. Ther. 2021, 11, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Nolan, H.R.; Wagner, M.L.; Jenkins, T.; Lim, F.-Y. Outcomes in the giant omphalocele population: A single center comprehensive experience. J. Pediatr. Surg. 2020, 55, 1866–1871. [Google Scholar] [CrossRef] [PubMed]
- Rees, C.M.; Tullie, L.; Pierro, A.; Kiely, E.; Curry, J.I.; Cross, K.; Yates, R.; Eaton, S.; De Coppi, P. Primary versus Staged Closure of Exomphalos Major: Cardiac Anomalies Do Not Affect Outcome. Eur. J. Pediatr. Surg. 2017, 28, 279–284, Erratum in Eur. J. Pediatr. Surg. 2018, 28, e1. [Google Scholar] [CrossRef] [PubMed]
- Elhedai, H.; Arul, G.S.; Yong, S.; Nagakumar, P.; Kanthimathinathan, H.K.; Jester, I.; Chaudhari, M.; Jones, T.J.; Stumper, O.; Seale, A.N. Outcomes of patients with exomphalos and associated congenital heart diseases. Pediatr. Surg. Int. 2022, 39, 12. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.F. Long-term Follow-up of “Simple” Lesions-Atrial Septal Defect, Ventricular Septal Defect, and Coarctation of the Aorta. Congenit. Heart Dis. 2015, 10, 466–474. [Google Scholar] [CrossRef]
- Connor, J.A.; Thiagarajan, R. Hypoplastic left heart syndrome. Orphanet J. Rare Dis. 2007, 2, 23. [Google Scholar] [CrossRef]
- Hainstock, M.; Bardo, D.; Rice, M.; Langley, S. Hypoplastic Left Heart Syndrome Associated with Large Omphalocele and Hypoplastic Left Bronchus. Pediatr. Cardiol. 2010, 31, 878–880. [Google Scholar] [CrossRef]
- Berg, C.; Lachmann, R.; Kaiser, C.; Kozlowski, P.; Stressig, R.; Schneider, M.; Asfour, B.; Herberg, U.; Breuer, J.; Gembruch, U.; et al. Prenatal diagnosis of tricuspid atresia: Intrauterine course and outcome. Ultrasound Obstet. Gynecol. 2010, 35, 183–190. [Google Scholar] [CrossRef]
- Kon, A.A.; Patel, A.; Leuthner, S.; Lantos, J.D. Parental Refusal of Surgery in an Infant With Tricuspid Atresia. Pediatrics 2016, 138, e20161730. [Google Scholar] [CrossRef]
- Pius, S.; Ibrahim, H.A.; Bello, M.; Tahir, M.B. Complete Ectopia Cordis: A Case Report and Literature Review. Case Rep. Pediatr. 2017, 2017, 1858621. [Google Scholar] [CrossRef] [PubMed]
- Alshamiri, K.M.; Albriek, A.Z.; Farrag, T.W.; Alshamiri, M.Q. Ectopia cordis in an adult patient with COVID-19: A case report and literature review. Clin. Case Rep. 2022, 10, e05389. [Google Scholar] [CrossRef]
- Mlczoch, E.; Carvalho, J.S. Interrupted inferior vena cava in fetuses with omphalocele. Case series of fetuses referred for fetal echocardiography and review of the literature. Early Hum. Dev. 2015, 91, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Danzer, E.; Edgar, J.C.; Eppley, E.; Goldshore, M.A.; Chotzoglou, E.; Herkert, L.M.; Oliver, E.R.; Rintoul, N.E.; Panitch, H.; Adzick, N.S.; et al. Predicting neonatal outcomes in infants with giant omphalocele using prenatal magnetic resonance imaging calculated observed-to-expected fetal lung volumes. Prenat. Diagn. 2021, 41, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Col, A.K.D.; Bhombal, S.; Tacy, T.A.; Hintz, S.R.; Feinstein, J.; Altit, G. Comprehensive Echocardiographic Assessment of Ventricular Function and Pulmonary Pressure in the Neonatal Omphalocele Population. Am. J. Perinatol. 2020, 38, e109–e115. [Google Scholar] [CrossRef]
- Liu, T.-X.; Du, L.-Z.; Ma, X.-L.; Chen, Z.; Shi, L.-P. Giant omphalocele associated pulmonary hypertension: A retrospective study. Front. Pediatr. 2022, 10, 940289. [Google Scholar] [CrossRef]
- Chen, C.-P. Syndromes and Disorders Associated with Omphalocele (I): Beckwith–Wiedemann Syndrome. Taiwan J. Obstet. Gynecol. 2007, 46, 96–102. [Google Scholar] [CrossRef]
- Bedei, I.; Gloning, K.; Joyeux, L.; Meyer-Wittkopf, M.; Willner, D.; Krapp, M.; Scharf, A.; Degenhardt, J.; Heling, K.; Kozlowski, P.; et al. Turner syndrome-omphalocele association: Incidence, karyotype, phenotype and fetal outcome. Prenat. Diagn. 2023, 43, 183–191. [Google Scholar] [CrossRef]
- Stoll, C.; Alembik, Y.; Dott, B.; Roth, M.-P. Omphalocele and gastroschisis and associated malformations. Am. J. Med. Genet. A 2008, 146A, 1280–1285. [Google Scholar] [CrossRef]
- Kim, J.B.; Park, J.-J.; Ko, J.K.; Goo, H.W.; Kim, Y.H.; Park, I.S.; Yun, T.J.; Seo, D.M. A case of PAGOD syndrome with hypoplastic left heart syndrome. Int. J. Cardiol. 2007, 114, 270–271. [Google Scholar] [CrossRef]
- Rossetti, L.Z.; Glinton, K.; Yuan, B.; Liu, P.; Pillai, N.; Mizerik, E.; Magoulas, P.; Rosenfeld, J.A.; Karaviti, L.; Sutton, V.R.; et al. Review of the phenotypic spectrum associated with haploinsufficiency of MYRF. Am. J. Med. Genet. Part A 2019, 179, 1376–1382. [Google Scholar] [CrossRef]
- Kennerknecht, I.; Sorgo, W.; Oberhoffer, R.; Teller, W.M.; Mattfeldt, T.; Negri, G.; Vogel, W. Familial occurrence of agonadism and multiple internal malformations in phenotypically normal girls with 46,XY and 46,XX karyotypes, respectively: A new autosomal recessive syndrome. Am. J. Med. Genet. 1993, 47, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Gil, L.; Sanchez-De-Toledo, J.; Ferreres, J.C.; Vendrell, T.; Ruiz-Campillo, C.; Balcells, J. Defecto diafragmático, cardiopatía congénita, agonadismo: Un nuevo caso de síndrome de PAGOD [Diaphragmatic defect, congenital heart disease, agonadism: A new case of PAGOD syndrome]. An. Pediatr. (Barc.) 2014, 81, e34–e35. [Google Scholar] [CrossRef] [PubMed]
- Herman, T.E.; McAlister, W.H.; Stazzone, M.M. PAGOD Syndrome: A New Abdominal Finding and Risk of Sudden Death. J. Perinatol. 2005, 25, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Skarsgard, E.D. Immediate versus staged repair of omphaloceles. Semin. Pediatr. Surg. 2019, 28, 89–94. [Google Scholar] [CrossRef]
- Weksberg, R.; Shuman, C.; Beckwith, J.B. Beckwith–Wiedemann syndrome. Eur. J. Hum. Genet. 2010, 18, 8. [Google Scholar] [CrossRef]
- Mussa, A.; Molinatto, C.; Cerrato, F.; Palumbo, O.; Carella, M.; Baldassarre, G.; Carli, D.; Peris, C.; Riccio, A.; Ferrero, G.B. Assisted Reproductive Techniques and Risk of Beckwith-Wiedemann Syndrome. Pediatrics 2017, 140, e20164311. [Google Scholar] [CrossRef]
- Shuman, C.; Beckwith, J.B.; Weksberg, R. Beckwith-Wiedemann Syndrome. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Eds.; University of Washington: Seattle, WA, USA, 2000. [Google Scholar]
- Williams, A.P.; Marayati, R.; Beierle, E.A. Pentalogy of Cantrell. Semin. Pediatr. Surg. 2019, 28, 106–110. [Google Scholar] [CrossRef]
- Sana, M.K.; Rentea, R.M. Pentalogy of Cantrell. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Cantrell, J.R.; Haller, J.A.; Ravitch, M.M. A syndrome of congenital defects involving the abdominal wall, sternum, diaphragm, pericardium, and heart. Surg. Gynecol. Obstet. 1958, 107, 602–614. [Google Scholar]
- Toyama, W.M. Combined congenital defects of the anterior abdominal wall, sternum, diaphragm, pericardium, and heart: A case report and review of the syndrome. Pediatrics 1972, 50, 778–792. [Google Scholar]
- Boe, N.M.; Rhee-Morris, L.; Towner, D.; Moon-Grady, A.J. Prenatal diagnosis of omphalocele and left atrial isomerism (polysplenia) including complex congenital heart disease with ventricular noncompaction cardiomyopathy. J. Ultrasound Med. 2008, 27, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Cheang, I.N.; Fu, Y.-W.; Chin, T.-W.; Hsu, Y.-J.; Wu, C.-Y. Splenic Torsion in Heterotaxy Syndrome with Left Isomerism: A Case Report and Literature Review. Diagnostics 2022, 12, 2920. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.C.; Greenbaum, B.; Hall, B.D. The OEIS complex (omphalocele, exstrophy, imperforate anus, spinal defects). Birth Defects Orig. Artic. Ser. 1978, 14, 253–263. [Google Scholar] [PubMed]
- Chen, C.-P. Syndromes and Disorders Associated with Omphalocele (II): OEIS Complex and Pentalogy Of Cantrell. Taiwan J. Obstet. Gynecol. 2007, 46, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Suzumori, N.; Obayashi, S.; Mizutani, E.; Hayashi, Y.; Sugiura-Ogasawara, M. Prenatal findings of omphalocele-exstrophy of the bladder-imperforate anus-spinal defects (OEIS) complex. Congenit. Anom. 2012, 52, 179–181. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Xue, Y.; Guo, Y. Prenatal ultrasound-based diagnosis of fetal OEIS complex associated with lower limb polymelia and cardiac, hepatic dysplasia: A case report. Clin. Case Rep. 2019, 7, 2153–2155. [Google Scholar] [CrossRef]
- Van Allen, M.I.; Curry, C.; Gallagher, L. Limb body wall complex: I. Pathogenesis. Am. J. Med. Genet. 1987, 28, 529–548. [Google Scholar] [CrossRef]
- Hartwig, N.G.; Vermeij-Keers, C.; De Vries, H.E.; Kagie, M.; Kragt, H. Limb body wall malformation complex: An embryologic etiology? Hum. Pathol. 1989, 20, 1071–1077. [Google Scholar] [CrossRef]
- Chikkannaiah, P.; Dhumale, H.; Kangle, R.; Shekar, R. Limb Body Wall Complex: A Rare Anomaly. J. Lab. Physicians 2013, 5, 65–67. [Google Scholar] [CrossRef]
- Hunter, A.G.; Seaver, L.H.; Stevenson, R.E. Limb-body wall defects. Is there a defensible hypothesis and can it explain all the associated anomalies? Am. J. Med. Genet. 2011, 155A, 2045–2059. [Google Scholar] [CrossRef]
- Seleim, H.M.; Wishahy, A.M.; Magdy, B.; Elseoudi, M.; Zakaria, R.H.; Kaddah, S.N.; Elbarbary, M.M. The dilemma after an unforeseen aortic arch anomalies at thoracoscopic repair of esophageal atresia: Is curtailing surgery still a necessity? Scand. J. Surg. 2022, 111, 14574969221090487. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Pujabet, M.; Gahete, J.A.M.; Guillén, G.; López-Fernández, S.; Martin-Giménez, M.P.; Lloret, J.; López, M. Management of neonates with right-sided aortic arch and esophageal atresia: International survey on IPEG AND ESPES members’ experience. J. Pediatr. Surg. 2018, 53, 1923–1927. [Google Scholar] [CrossRef] [PubMed]
- Czichos, E.; Lukaszek, S.; Krekora, M.; Kulig, A.; Wilczyński, J. Early amnion rupture and fetal and newborn defects as an obstetrical and pathomorphological problem. Ginekol. Pol. 2005, 76, 448–456. [Google Scholar]
- Naudion, S.; Moutton, S.; Coupry, I.; Solé, G.; Deforges, J.; Guerineau, E.; Hubert, C.; Deves, S.; Pilliod, J.; Rooryck, C.; et al. Fetal phenotypes in otopalatodigital spectrum disorders. Clin. Genet. 2015, 89, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.J.; Flyer, J.N.; Edwards, E.M.; Soll, R.F.; Horbar, J.D.; Yeager, S.B. Outcomes for Ectopia Cordis. J. Pediatr. 2020, 216, 67–72. [Google Scholar] [CrossRef]
- Karamlou, T.; Gurofsky, R.; Al Sukhni, E.; Coles, J.G.; Williams, W.G.; Caldarone, C.A.; Van Arsdell, G.S.; McCrindle, B.W. Factors Associated With Mortality and Reoperation in 377 Children with Total Anomalous Pulmonary Venous Connection. Circulation 2007, 115, 1591–1598. [Google Scholar] [CrossRef]
- Ţarcă, E.; Roșu, S.T.; Cojocaru, E.; Trandafir, L.; Luca, A.C.; Lupu, V.V.; Moisă, M.; Munteanu, V.; Butnariu, L.I.; Ţarcă, V. Statistical Analysis of the Main Risk Factors of an Unfavorable Evolution in Gastroschisis. J. Pers. Med. 2021, 11, 1168. [Google Scholar] [CrossRef]
- Adams, A.D.; Stover, S.; Rac, M.W. Omphalocele—What should we tell the prospective parents? Prenat. Diagn. 2021, 41, 486–496. [Google Scholar] [CrossRef]
- Anandakumar, C.; Badruddin, M.N.; Chua, T.M.; Wong, Y.C.; Chia, D. First-trimester prenatal diagnosis of omphalocele using three-dimensional ultrasonography. Ultrasound Obstet. Gynecol. 2002, 20, 635–636. [Google Scholar] [CrossRef]
- Mozumdar, N.; Rowland, J.; Pan, S.; Rajagopal, H.; Geiger, M.K.; Srivastava, S.; Stern, K.W. Diagnostic Accuracy of Fetal Echocardiography in Congenital Heart Disease. J. Am. Soc. Echocardiogr. 2020, 33, 1384–1390. [Google Scholar] [CrossRef]
- Engels, A.C.; Debeer, A.; Russo, F.M.; Aertsen, M.; Aerts, K.; Miserez, M.; Deprest, J.; Lewi, L.; Devlieger, R. Pericardio-Amniotic Shunting for Incomplete Pentalogy of Cantrell. Fetal Diagn. Ther. 2017, 41, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Partridge, E.A.; Hanna, B.D.; Panitch, H.B.; Rintoul, N.E.; Peranteau, W.H.; Flake, A.W.; Scott Adzick, N.; Hedrick, H.L. Pulmonary hypertension in giant om- phalocele infants. J. Pediatr. Surg. 2014, 49, 1767–1770. [Google Scholar] [CrossRef] [PubMed]
- Nasr, A.; McNamara, P.; Mertens, L.; Levin, D.; James, A.; Holtby, H.; Langer, J.C. Is routine preoperative 2-dimensional echocardiography necessary for infants with esophageal atresia, omphalocele, or anorectal malformations? J. Pediatr. Surg. 2010, 45, 876–879. [Google Scholar] [CrossRef] [PubMed]
- Duceac, L.D.; Marcu, C.; Ichim, D.L.; Ciomaga, I.M.; Tarca, E.; Iordache, A.C.; Ciuhodaru, M.I.; Florescu, L.; Tutunaru, D.; Luca, A.C.; et al. Antibiotic Molecules Involved in Increasing Microbial Resistance. Rev. Chim. 2019, 70, 2622–2626. [Google Scholar] [CrossRef]
- Moroşan, E.; Mihailovici, M.S.; Giuşcă, S.E.; Cojocaru, E.; Avădănei, E.R.; Căruntu, I.D.; Teleman, S. Hepatic steatosis background in chronic hepatitis B and C—significance of similarities and differences. Rom. J. Morphol. Embryol. 2014, 55, 1041–1047. [Google Scholar]
- Cohen, J.L.; Cielo, C.M.; Kupa, J.; Duffy, K.A.; Hathaway, E.R.; Kalish, J.M.; Taylor, J.A. The Utility of Early Tongue Reduction Surgery for Macroglossia in Beckwith-Wiedemann Syndrome. Plast. Reconstr. Surg. 2020, 145, 803e–813e. [Google Scholar] [CrossRef]
- Tirrell, T.F.; Demehri, F.R.; Henry, O.S.; Cullen, L.; Lillehei, C.W.; Warf, B.C.; Gates, R.L.; Borer, J.G.; Dickie, B.H. Safety of delayed surgical repair of omphalocele–exstrophy–imperforate anus–spinal defects (OEIS) complex in infants with significant comorbidities. Pediatr. Surg. Int. 2020, 37, 93–99. [Google Scholar] [CrossRef]
- Saxena, A.K.; Van Tuil, C. Delayed three-stage closure of giant omphalocele using pericard patch. Hernia 2007, 12, 201–203. [Google Scholar] [CrossRef]
- Zhang, X.; Xing, Q.; Sun, J.; Hou, X.; Kuang, M.; Zhang, G. Surgical treatment and outcomes of pentalogy of Cantrell in eight patients. J. Pediatr. Surg. 2014, 49, 1335–1340. [Google Scholar] [CrossRef]
- Divkovic, D.; Kvolik, S.; Sipl, M.; Sego, K.; Pušeljić, S.; Rakipovic-Stojanovic, A.; Kovačić, B. A successful early gore-tex reconstruction of an abdominal wall defect in a neonate with Cantrell pentalogy: A case report and literature review. Clin. Case Rep. 2014, 3, 19–23. [Google Scholar] [CrossRef]
- Naren Satya, S.M.; Mayilvaganan, K.R.; Prathyusha, I.S.; Gautam, M.S.; Raidu, D.; Amogh, V.N. A Recurrent Case of Pentalogy of Cantrell: A Rare Case with Sonological Findings and Review of Literature. Pol. J. Radiol. 2017, 82, 28–31. [Google Scholar] [CrossRef]
- Nembhard, W.N.; Bergman, J.E.H.; Politis, M.D.; Arteaga-Vázquez, J.; Bermejo-Sánchez, E.; Canfield, M.A.; Cragan, J.D.; Dastgiri, S.; De Walle, H.E.K.; Feldkamp, M.L.; et al. A multi-country study of prevalence and early childhood mortality among children with omphalocele. Birth Defects Res. 2020, 112, 1787–1801. [Google Scholar] [CrossRef] [PubMed]
- Fogelström, A.; Caldeman, C.; Oddsberg, J.; Granström, A.L.; Burgos, C.M. Omphalocele: National current birth prevalence and survival. Pediatr. Surg. Int. 2021, 37, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Horodnic, A.V.; Apetrei, A.; Luca, F.-A.; Ciobanu, C.-I. Rating healthcare services: Consumer satisfaction vs. health system performance. Serv. Ind. J. 2018, 38, 974–994. [Google Scholar] [CrossRef]
- Xu, S.-T.; Zhang, G.-L.; Zhang, X.-M.; Huang, Z.-J.; Mei, S.-S.; Zhong, W.; Li, D.-Z. Can perinatal outcomes of fetal omphalocele be improved at a tertiary center in South China? J. Matern. Neonatal Med. 2021, 35, 8409–8411. [Google Scholar] [CrossRef]
- Barrios-Sanjuanelo, A.; Abelló-Munarriz, C.; Cardona-Arias, J.A. Mortality in neonates with giant omphalocele subjected to a surgical technique in Barranquilla, Colombia from 1994 to 2019. Sci. Rep. 2021, 11, 310. [Google Scholar] [CrossRef]


{kind=link}
| Cardiac Defect | Incidence | Mortality | Syndromes or Chromosomal Abnormalities |
|---|---|---|---|
| Atrial septal defects | 1.3 per 1000 live births | Less than 1% | Trisomy 13, 18, and Down syndrome, Turner syndrome, PAGOD syndrome, VACTERL malformation association |
| Ventricular septal defect (VSD) | 4.2 per 1000 live births | Surgically closed VSD displayed mortality rates of 1% and up to 3% if left untreated | Trisomy 13, 18, 21, Turner syndrome, PAGOD syndrome, Limb body wall complex, Pentalogy of Cantrell |
| Hypoplastic left heart syndrome | 0.016 to 0.036% of all live births | About 20% to 60% of babies survive their first year of life | Turner syndrome, PAGOD syndrome |
| Tricuspid atresia | 0.1 per 1000 live birth | The mortality rate after birth is up to 11% in the neonatal period and 40% after 20 years | VACTERL malformation association, Limb body wall complex |
| Ectopia cordis | 5.5 to 7.9/million live births | Mortality exceeded 50% for infants <2500 g and <37 weeks of gestation. | Pentalogy of Cantrell, Limb body wall complex |
| Associated abnormalities of systemic veins | 0.3% to 0.4% of the general population | Mortality up to 40%, depending on the type of abnormalities and associated defects | ADAM sequence, left atrial isomerism |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Țarcă, E.; Al Namat, D.; Luca, A.C.; Lupu, V.V.; Al Namat, R.; Lupu, A.; Bălănescu, L.; Bernic, J.; Butnariu, L.I.; Moscalu, M.; et al. Omphalocele and Cardiac Abnormalities—The Importance of the Association. Diagnostics 2023, 13, 1413. https://doi.org/10.3390/diagnostics13081413
Țarcă E, Al Namat D, Luca AC, Lupu VV, Al Namat R, Lupu A, Bălănescu L, Bernic J, Butnariu LI, Moscalu M, et al. Omphalocele and Cardiac Abnormalities—The Importance of the Association. Diagnostics. 2023; 13(8):1413. https://doi.org/10.3390/diagnostics13081413
Chicago/Turabian StyleȚarcă, Elena, Dina Al Namat, Alina Costina Luca, Vasile Valeriu Lupu, Razan Al Namat, Ancuța Lupu, Laura Bălănescu, Jana Bernic, Lăcrămioara Ionela Butnariu, Mihaela Moscalu, and et al. 2023. "Omphalocele and Cardiac Abnormalities—The Importance of the Association" Diagnostics 13, no. 8: 1413. https://doi.org/10.3390/diagnostics13081413