
Detection of Mosaic Absence of Heterozygosity (AOH) Using Low-Pass Whole Genome Sequencing in Prenatal Diagnosis: A Preliminary Report
Abstract
:1. Introduction
2. Materials and Methods
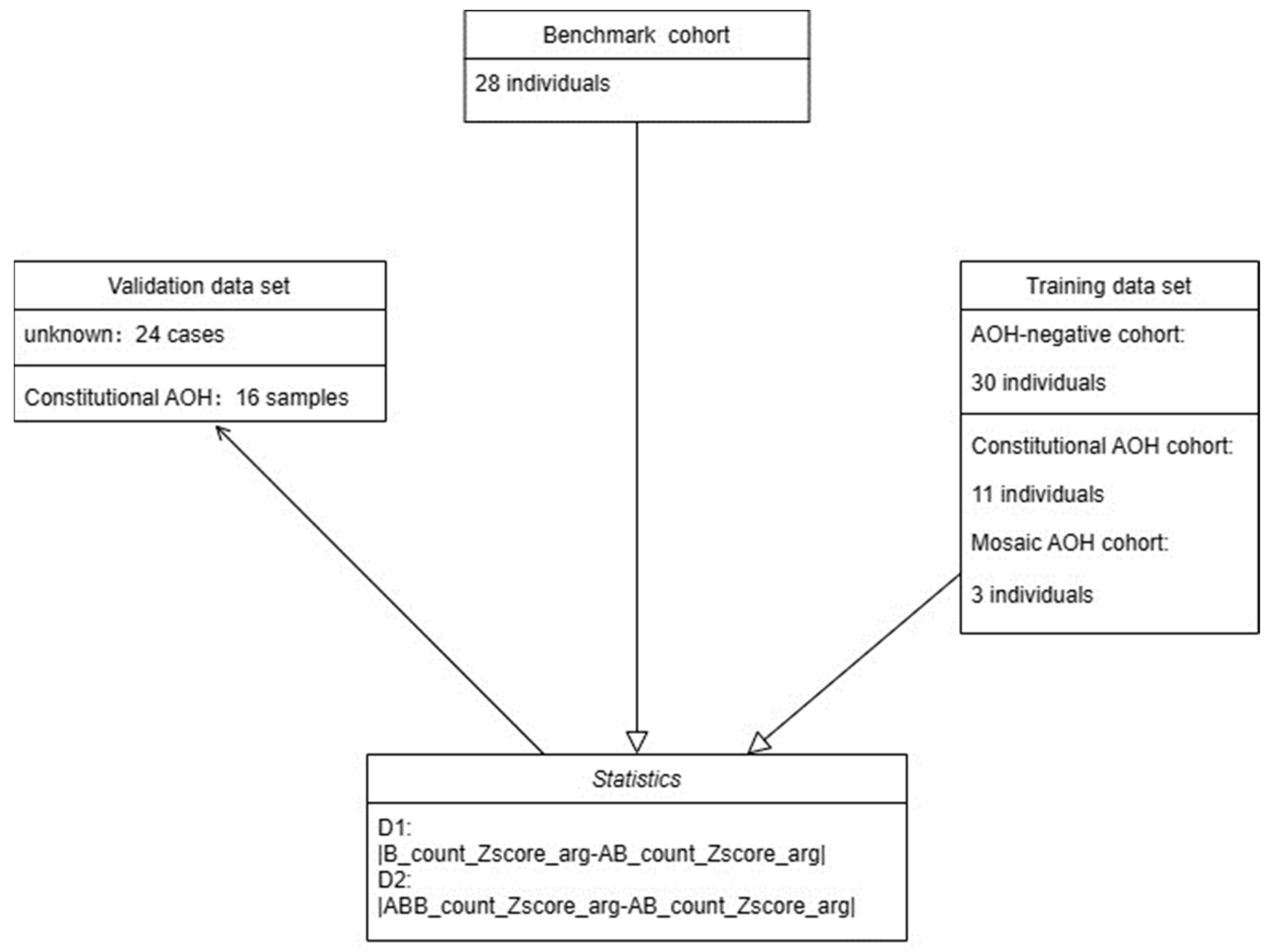
2.1. Study Design and Sample Preparation
2.2. Samples
2.3. Methods
2.3.1. Sequencing, Quality Control, and Variant Calling
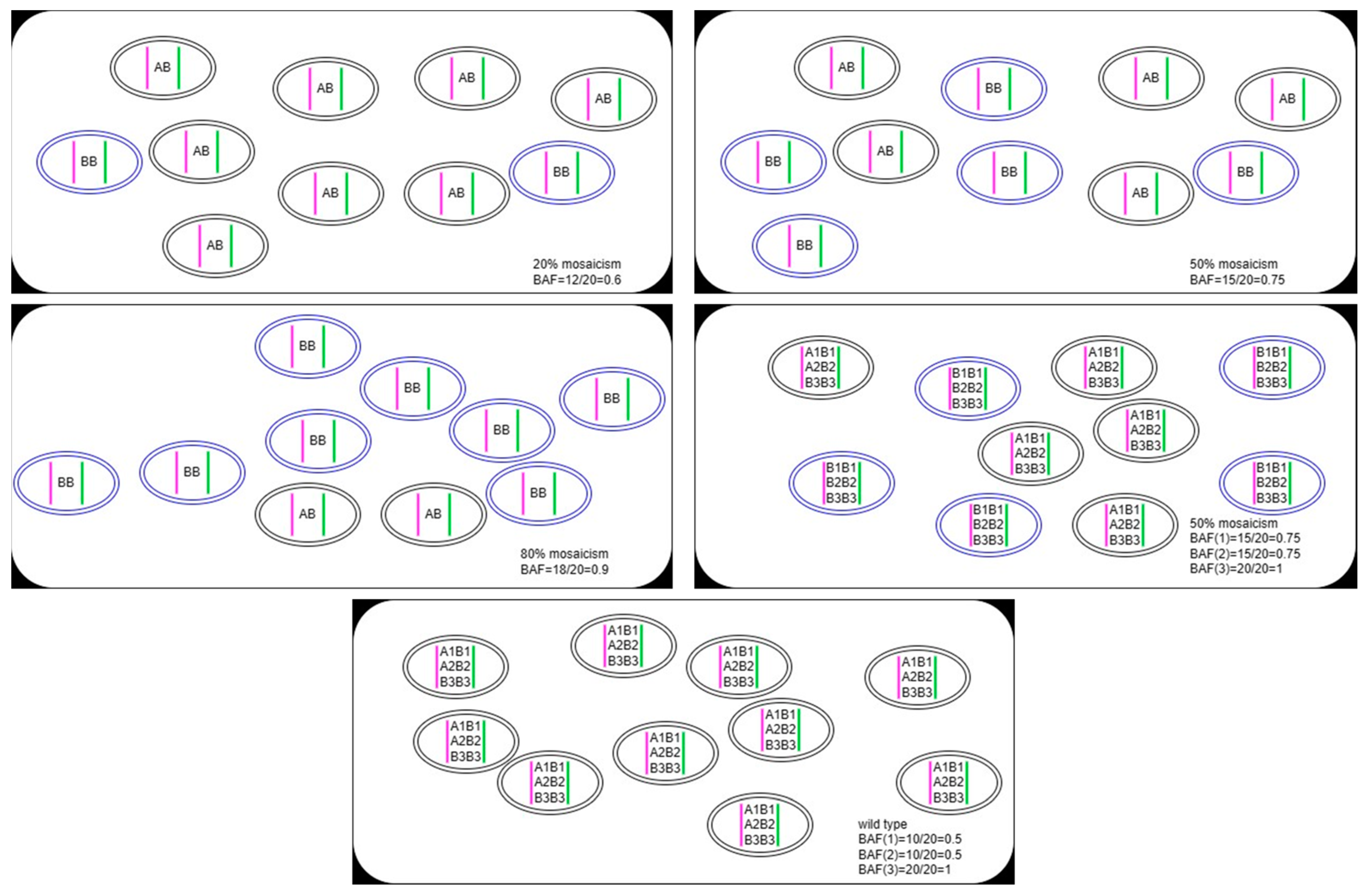
2.3.2. Genotype Status
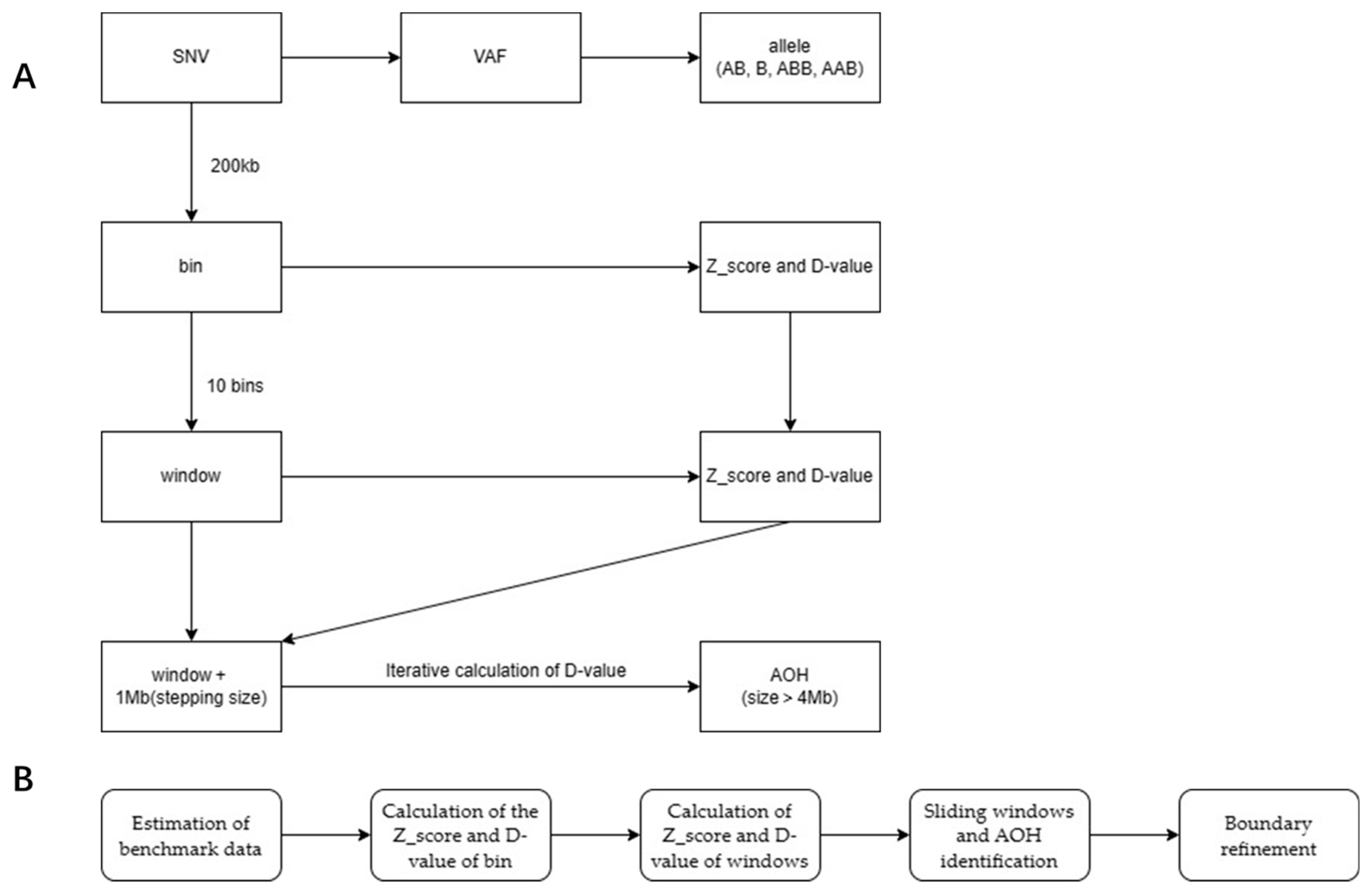
2.3.3. Bioinformatics Analysis
2.3.4. Chromosomal Microarray Analysis
3. Results
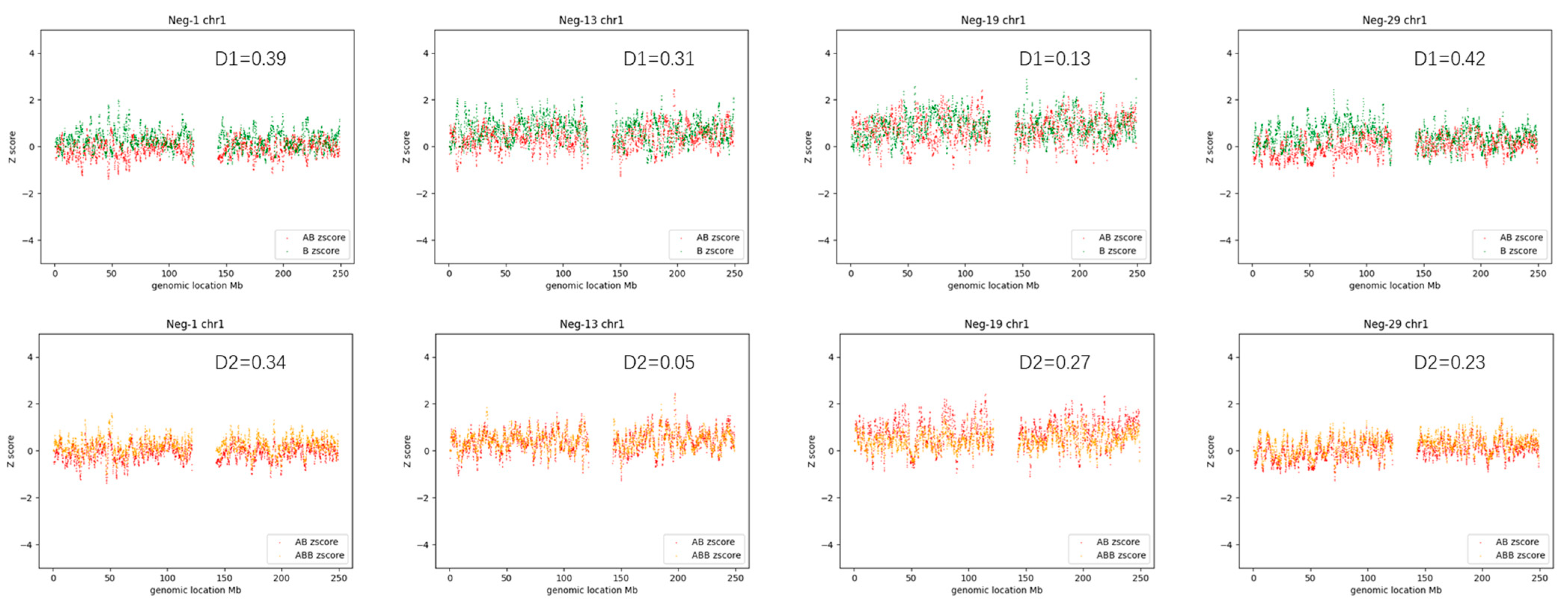
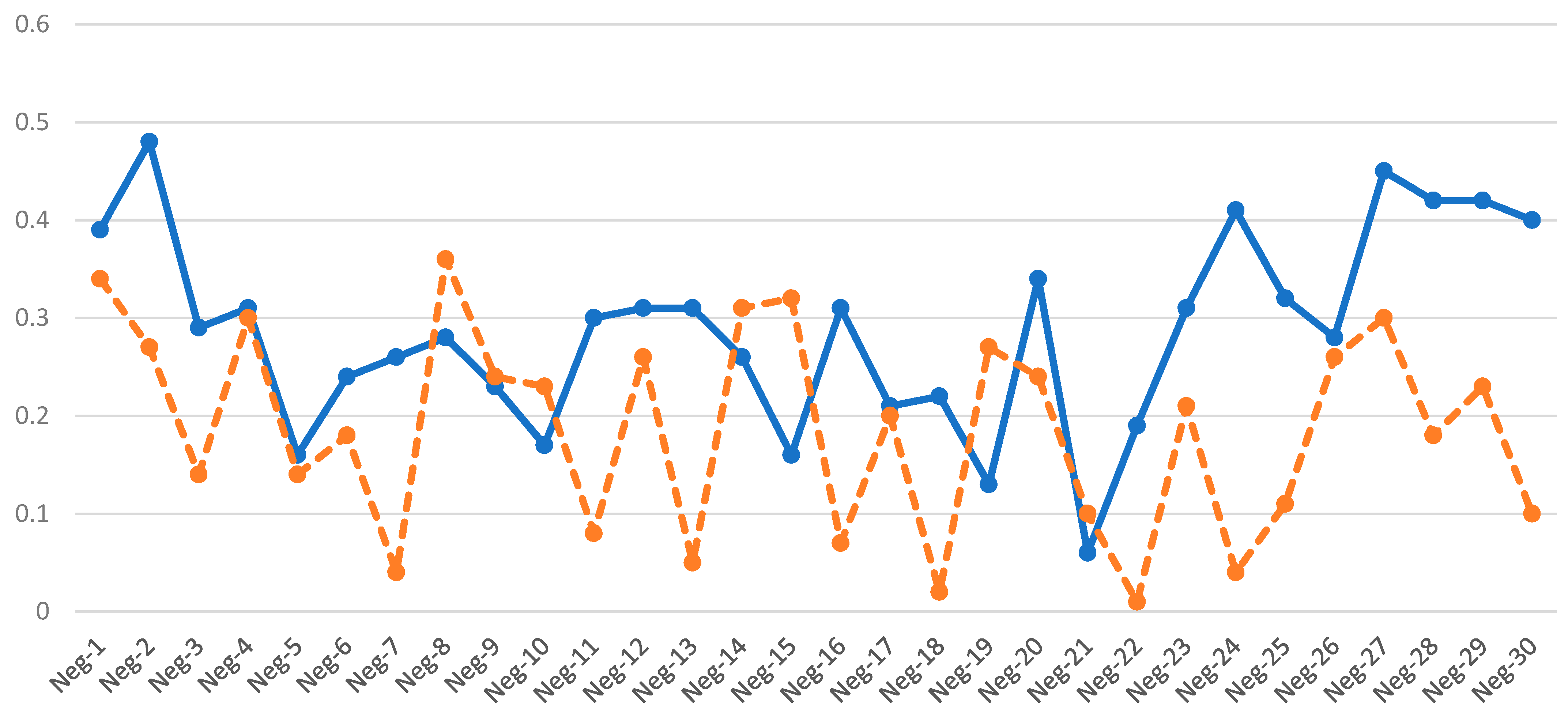
3.1. AOH-Negative Samples
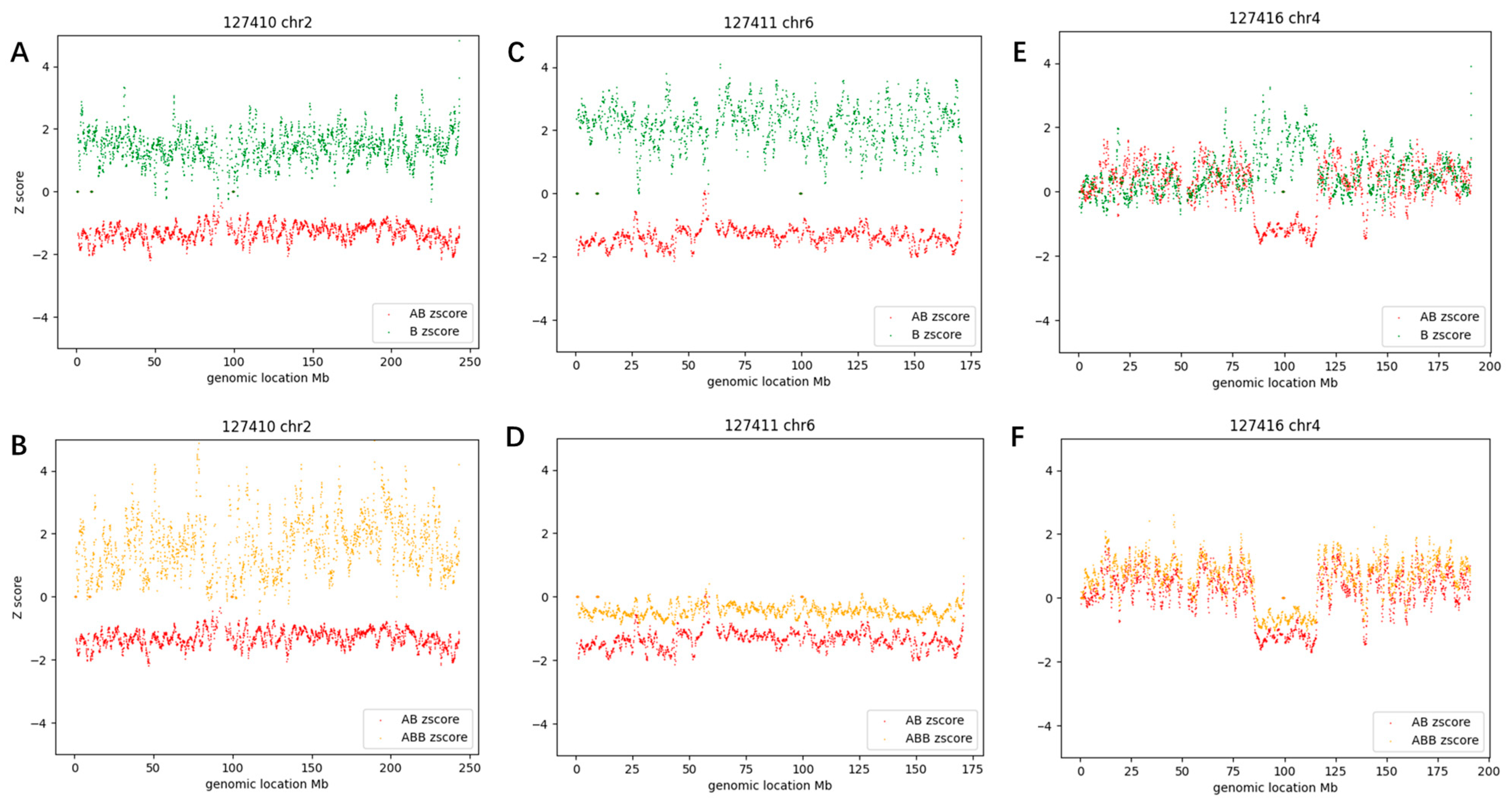
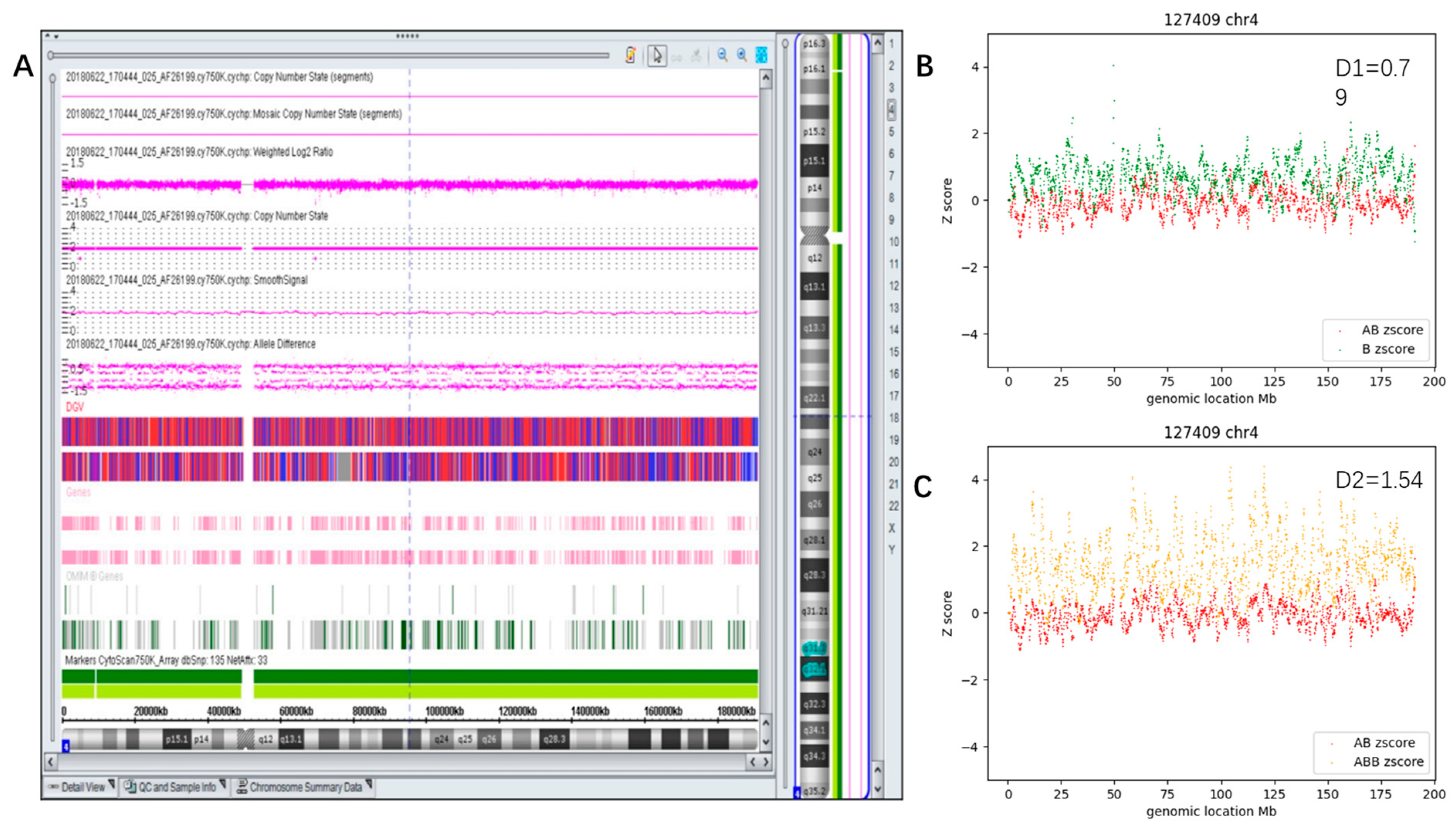
3.2. Constitutional AOH Samples
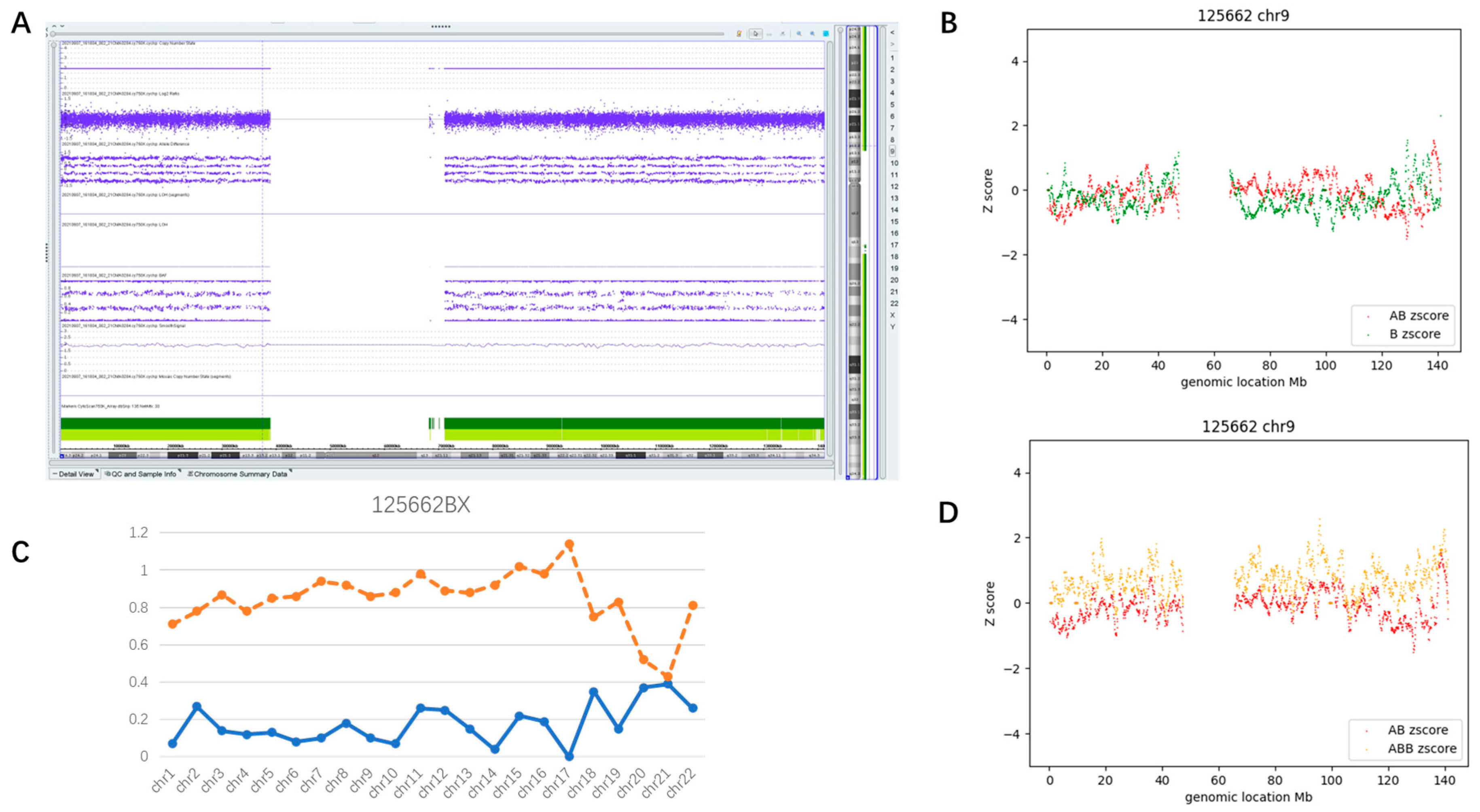
3.3. Mosaic AOH Cases
3.3.1. Whole Genome Mosaic AOH (Case #28)
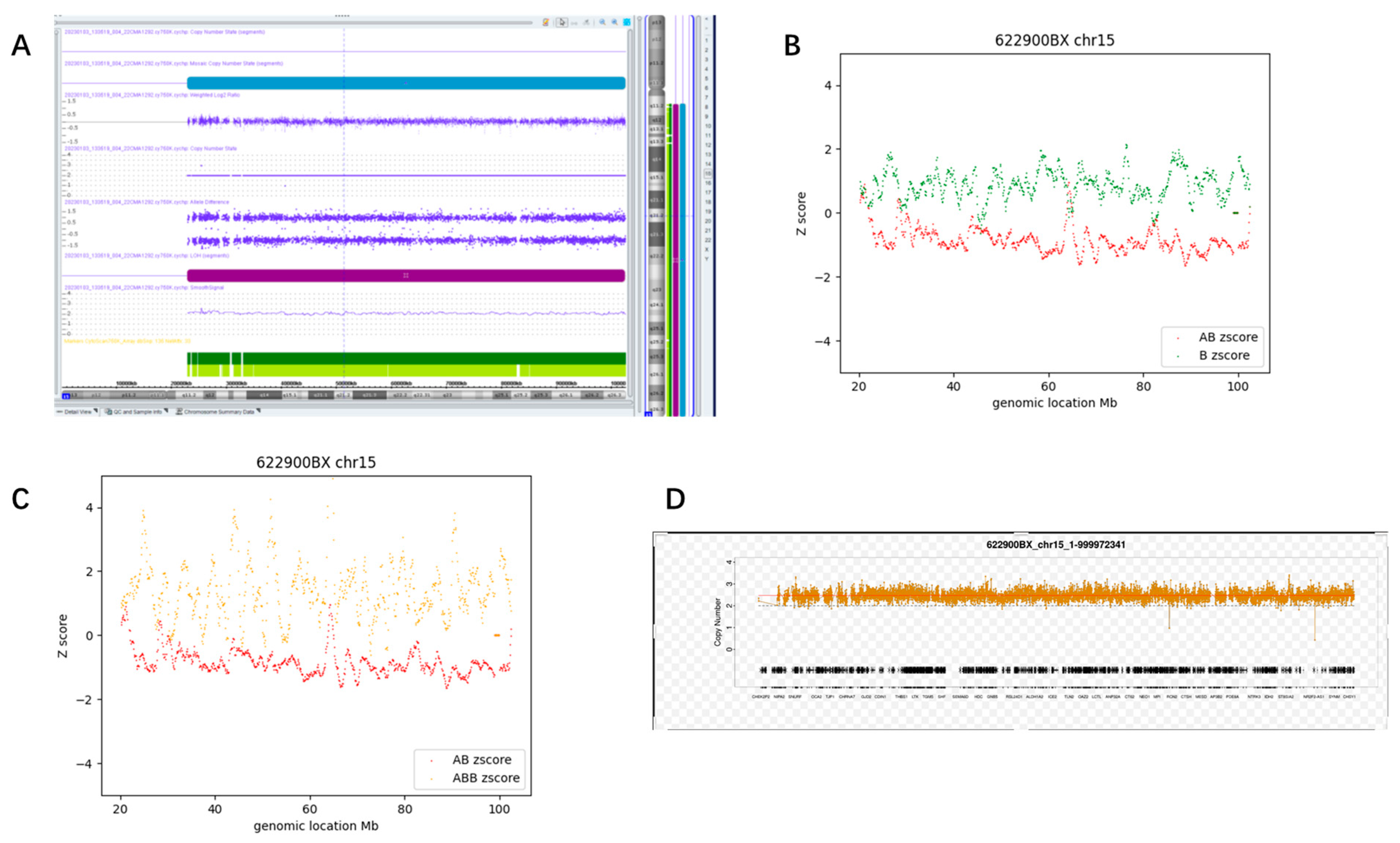
3.3.2. Whole Chromosome Mosaic AOH (Case #60)
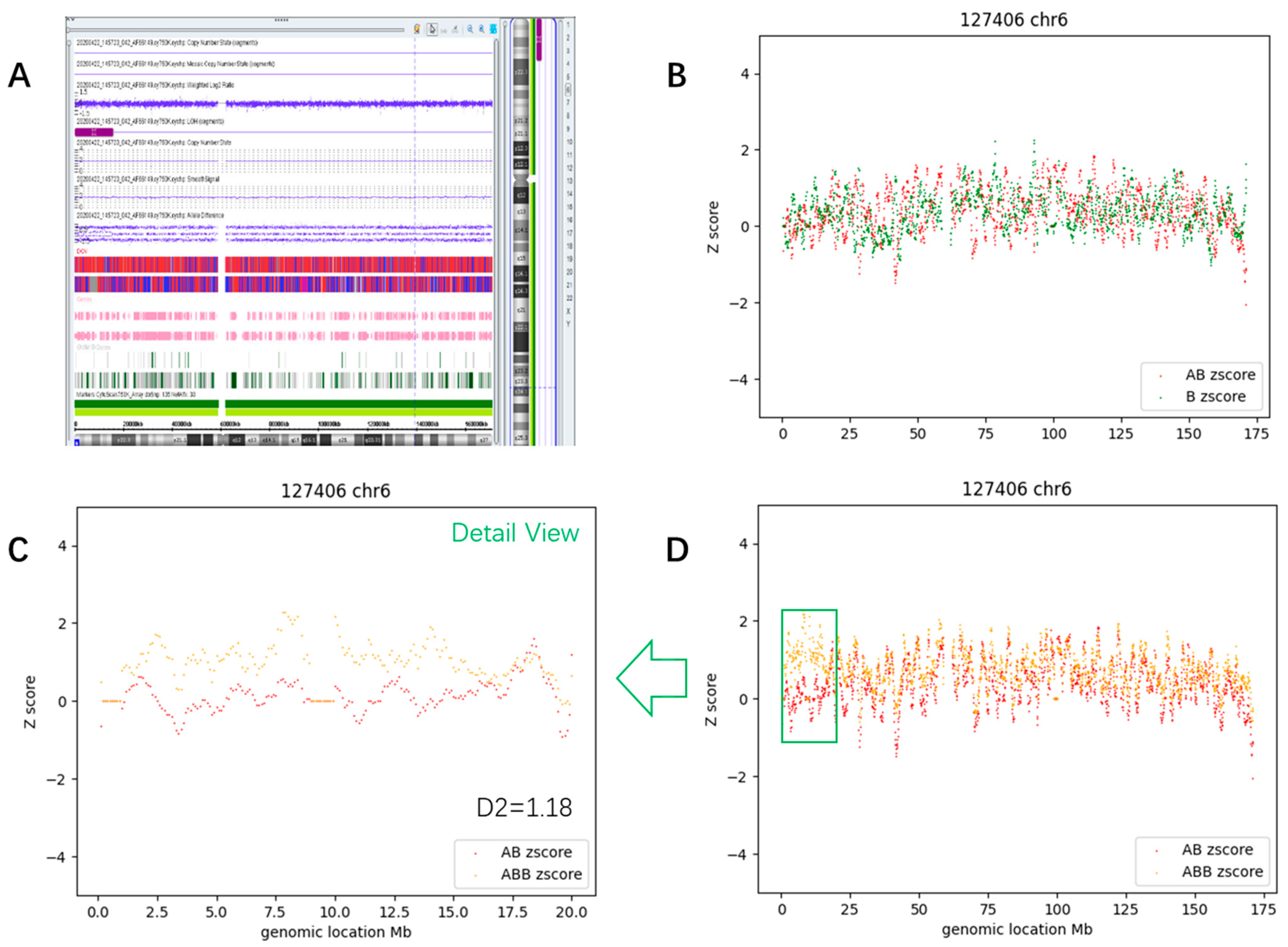
3.3.3. Segmental Mosaic AOH (Case #63)
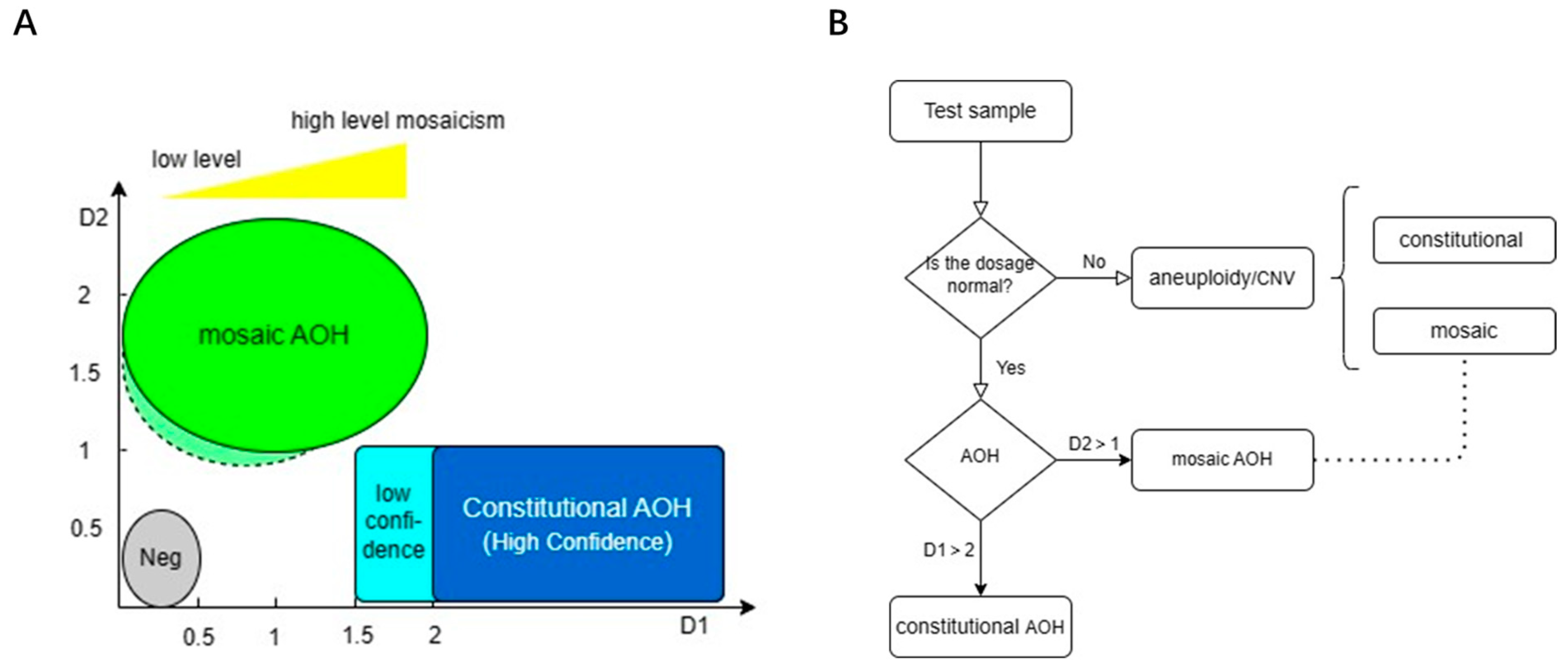
3.4. Determination of Thresholds and Detection Pipeline
3.5. Validation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Biesecker, L.G.; Spinner, N.B. A genomic view of mosaicism and human disease. Nat. Rev. Genet. 2013, 14, 307–320. [Google Scholar] [CrossRef]
- Eggermann, T.; Soellner, L.; Buiting, K.; Kotzot, D. Mosaicism and uniparental disomy in prenatal diagnosis. Trends Mol. Med. 2015, 21, 77–87. [Google Scholar] [CrossRef]
- Stern, C. Somatic crossing over and segregation in Drosophila melanogaster. Genetics 1936, 21, 625–730. [Google Scholar] [CrossRef]
- Engel, E. A new genetic concept: Uniparental disomy and its potential effect, isodisomy. Am. J. Med. Genet. 1980, 6, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.E.; Perciaccante, R.G.; Greig, G.M.; Willard, H.F.; Ledbetter, D.H.; Hejtmancik, J.F.; Pollack, M.S.; O’Brien, W.E.; Beaudet, A. Uniparental disomy as a mechanism for human genetic disease. Am. J. Hum. Genet. 1988, 42, 217–226. [Google Scholar] [PubMed]
- Dutta, A.; Dutreux, F.; Schacherer, J. Loss of heterozygosity results in rapid but variable genome homogenization across yeast genetic backgrounds. Elife 2021, 10, e70339. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, V.; Meneses, H.N.; Fernández, L.; Martínez-Glez, V.; Gracia-Bouthelier, R.; F Fraga, M.; Guillén, E.; Nevado, J.; Gean, E.; Martorell, L.; et al. Beckwith–Wiedemann syndrome and uniparental disomy 11p: Fine mapping of the recombination breakpoints and evaluation of several techniques. Eur. J. Hum. Genet. 2011, 19, 416–421. [Google Scholar] [CrossRef]
- Robinson, W.P.; Christian, S.L.; Kuchinka, B.D.; Peñaherrera, M.S.; Das, S.; Schuffenhauer, S.; Malcolm, S.; Schinzel, A.A.; Hassold, T.J.; Ledbetter, D.H. Somatic segregation errors predominantly contribute to the gain or loss of a paternal chromosome leading to uniparental disomy for chromosome 15. Clin. Genet. 2000, 57, 349–358. [Google Scholar] [CrossRef]
- Tuna, M.; Knuutila, S.; Mills, G.B. Uniparental disomy in cancer. Trends Mol. Med. 2009, 15, 120–128. [Google Scholar] [CrossRef]
- Robinson, W.P. Mechanisms leading to uniparental disomy and their clinical consequences. Bioessays 2000, 22, 452–459. [Google Scholar] [CrossRef]
- Kaiser-Rogers, K.A.; Mcfadden, D.E.; Livasy, C.A.; Dansereau, J.; Jiang, R.; Knops, J.F.; Lefebvre, L.; Rao, K.W.; Robinson, W.P. Androgenetic/biparental mosaicism causes placental mesenchymal dysplasia. J. Med. Genet. 2006, 43, 187–192. [Google Scholar] [CrossRef]
- Chen, J.-M.; Cooper, D.N.; Chuzhanova, N.; Férec, C.; Patrinos, G.P. Gene conversion: Mechanisms, evolution and human disease. Nat. Rev. Genet. 2007, 8, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Smukowski Heil, C.S. Loss of heterozygosity and its importance in genome evolution. J. Mol. Evol. 2023, 91, 369–377. [Google Scholar] [CrossRef]
- Obstetricians, A.C.; Genetics, G.C. Committee Opinion No. 581: The use of chromosomal microarray analysis in prenatal diagnosis. Obstet. Gynecol. 2013, 122, 1374–1377. [Google Scholar]
- Levy, B.; Burnside, R.D. Are all chromosome microarrays the same? What clinicians need to know. Prenat. Diagn. 2019, 39, 157–164. [Google Scholar] [CrossRef]
- Magi, A.; Tattini, L.; Palombo, F.; Benelli, M.; Gialluisi, A.; Giusti, B.; Abbate, R.; Seri, M.; Gensini, G.F.; Romeo, G.; et al. H3M2: Detection of runs of homozygosity from whole-exome sequencing data. Bioinformatics 2014, 30, 2852–2859. [Google Scholar] [CrossRef] [PubMed]
- Chaubey, A.; Shenoy, S.; Mathur, A.; Ma, Z.; Valencia, C.A.; Reddy Nallamilli, B.R.; Szekeres, E., Jr.; Stansberry, L.; Liu, R.; Hegde, M.R. Low-Pass Genome Sequencing: Validation and Diagnostic Utility from 409 Clinical Cases of Low-Pass Genome Sequencing for the Detection of Copy Number Variants to Replace Constitutional Microarray. J. Mol. Diagn. 2020, 22, 823–840. [Google Scholar] [CrossRef]
- Dong, Z.; Chau, M.H.K.; Zhang, Y.; Yang, Z.; Shi, M.; Wah, Y.M.; Kwok, Y.K.; Leung, T.Y.; Morton, C.C.; Choy, K.W. Low-pass genome sequencing-based detection of absence of heterozygosity: Validation in clinical cytogenetics. Genet. Med. Off. J. Am. Coll. Med. Genet. 2021, 23, 1225–1233. [Google Scholar] [CrossRef]
- Lü, Y.; Jiang, Y.; Zhou, X.; Hao, N.; Lü, G.; Guo, X.; Guo, R.; Liu, W.; Xu, C.; Chang, J.; et al. Evaluation and Analysis of Absence of heterozygosity (AOH) Using Chromosome Analysis by Medium Coverage Whole Genome Sequencing (CMA-seq) in Prenatal Diagnosis. Diagnostics 2023, 13, 560. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Santiago, B.; Malats, N.; Rothman, N.; Armengol, L.; Garcia-Closas, M.; Kogevinas, M.; Villa, O.; Hutchinson, A.; Earl, J.; Marenne, G.; et al. Mosaic uniparental disomies and aneuploidies as large structural variants of the human genome. Am. J. Hum. Genet. 2010, 87, 129–138. [Google Scholar] [CrossRef]
- Zhang, F.; Flickinger, M.; Taliun, S.A.; Abecasis, G.R.; Scott, L.J.; McCaroll, S.A.; Pato, C.N.; Boehnke, M.; Kang, H.M. Ancestry-agnostic estimation of DNA sample contamination from sequence reads. Genome Res. 2020, 30, 185–194. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Qi, Q.; Jiang, Y.; Zhou, X.; Meng, H.; Hao, N.; Chang, J.; Bai, J.; Wang, C.; Wang, M.; Guo, J.; et al. Simultaneous Detection of CNVs and SNVs Improves the Diagnostic Yield of Fetuses with Ultrasound Anomalies and Normal Karyotypes. Genes 2020, 11, 1397. [Google Scholar] [CrossRef] [PubMed]
- Berend, S.A.; Horwitz, J.; McCaskill, C.; Shaffer, L.G. Identification of uniparental disomy following prenatal detection of Robertsonian translocations and isochromosomes. Am. J. Hum. Genet. 2000, 66, 1787–1793. [Google Scholar] [CrossRef] [PubMed]
- Conlin, L.K.; Thiel, B.D.; Bonnemann, C.G.; Medne, L.; Ernst, L.M.; Zackai, E.H.; Deardorff, M.A.; Krantz, I.D.; Hakonarson, H.; Spinner, N.B.; et al. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum. Mol. Genet. 2010, 19, 1263–1275. [Google Scholar] [CrossRef]
- Dou, Y.; Kwon, M.; Rodin, R.E.; Cortés-Ciriano, I.; Doan, R.; Luquette, L.J.; Galor, A.; Bohrson, C.; Walsh, C.A.; Park, P.J.; et al. Accurate detection of mosaic variants in sequencing data without matched controls. Nat. Biotechnol. 2020, 38, 314–319. [Google Scholar] [CrossRef]
- del Gaudio, D.; Shinawi, M.; Astbury, C.; Tayeh, M.K.; Deak, K.L.; Raca, G. Diagnostic Testing for Uniparental Disomy: A Points to Consider Statement from the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2020, 22, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Meynert, A.M.; Ansari, M.; FitzPatrick, D.R.; Taylor, M.S. Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinform. 2014, 15, 247. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Subgroup | D1 | D2 |
|---|---|---|---|
| Negative | <0.5 | <0.6 | |
| AOH (mosaic) | Whole chromosome | <1.5 | >1 |
| Segmental (size > 10 Mb) | <1.5 | >1 | |
| Whole genome | <0.5 * | >0.85 * | |
| AOH (constitutional) | Whole chromosome | >2 (stringent); >1.5 (loose) | <1 |
| Segmental (size > 4 Mb) | >2 (stringent); >1.5 (loose) | <1 | |
| Whole genome | >2 * | >2 * |
| Case Code | CMA | Size (Mb) | CMA-Seq | Size (Mb) |
|---|---|---|---|---|
| #164 | arr[hg19] 1p35.2p33(31,316,029_47,996,204) ×2 hmz | 16.7 | pos-chr1:23,000,000–49,000,000 | 26 |
| pos-chr12:69,000,000–78,000,000 | 9 | |||
| #165 | arr[hg19] 15q11.2q26.3(22,817,871_102,397,317) ×2 hmz | 79.6 | pos-chr15:0–103,000,000 | Whole chr |
| #166 | arr[hg19] 10p13p11.21(13,817,396_36,384,851) ×2 hmz | 22.6 | pos-chr10:12,000,000–37,000,000 | 25 |
| pos-chr8:33,000,000–58,000,000 | 25 | |||
| #168 | arr[hg19] 18q21.31q23(55,513,044_77,997,606) ×2 hmz | 22.5 | pos-chr18:55,000,000–78,000,000 | 23 |
| #170 | arr[hg19] 12q15q21.31(70,806,679_83,527,548) ×2 hmz | 12.7 | ||
| #171 | arr[hg19] 4p16.2p15.32(5,771,557_15,985,454) ×2 hmz | 10.3 | pos-chr4:8,000,000–16,000,000 | 8 |
| arr[hg19] 6p22.3p21.1(15,751,330_41,796,817) ×2 hmz | 26 | pos-chr6:14,000,000–40,000,000 | 26 | |
| #172 | arr[hg19] 22q11.1q13.33(16,888,900_51,157,531) ×2 hmz | 34.3 | pos-chr22:0–52,000,000 | Whole chr |
| #173 | arr[hg19] 1q23.3q25.3(161,955,758_180,706,775) ×2 hmz | 18.7 | pos-chr1:160,000,000–185,000,000 | 25 |
| #167 | arr[hg19] 15q11.2q26.3(22,817,871_102,397,317) ×2–3 hmz | 79.6 | pos-chr15:0–103,000,000 | Whole chr |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lü, Y.; Jiang, Y.; Zhou, X.; Hao, N.; Xu, C.; Guo, R.; Chang, J.; Li, M.; Zhang, H.; Zhou, J.; et al. Detection of Mosaic Absence of Heterozygosity (AOH) Using Low-Pass Whole Genome Sequencing in Prenatal Diagnosis: A Preliminary Report. Diagnostics 2023, 13, 2895. https://doi.org/10.3390/diagnostics13182895
Lü Y, Jiang Y, Zhou X, Hao N, Xu C, Guo R, Chang J, Li M, Zhang H, Zhou J, et al. Detection of Mosaic Absence of Heterozygosity (AOH) Using Low-Pass Whole Genome Sequencing in Prenatal Diagnosis: A Preliminary Report. Diagnostics. 2023; 13(18):2895. https://doi.org/10.3390/diagnostics13182895
Chicago/Turabian StyleLü, Yan, Yulin Jiang, Xiya Zhou, Na Hao, Chenlu Xu, Ruidong Guo, Jiazhen Chang, Mengmeng Li, Hanzhe Zhang, Jing Zhou, and et al. 2023. "Detection of Mosaic Absence of Heterozygosity (AOH) Using Low-Pass Whole Genome Sequencing in Prenatal Diagnosis: A Preliminary Report" Diagnostics 13, no. 18: 2895. https://doi.org/10.3390/diagnostics13182895